Thrombocytopenia cause , clinical feature and management
Reading File
Finding Sources
Searching PubMed
thrombocytopenia management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive information to provide a thorough answer. Here is the complete structured guide:
Thrombocytopenia: Causes, Clinical Features, and Management
Definition
Thrombocytopenia is defined as a platelet count below the normal range - typically <140,000-150,000/μL. However, clinically significant bleeding risk correlates with count thresholds:
| Platelet Count | Clinical Significance |
|---|---|
| 100,000-150,000/μL | Usually asymptomatic; mild risk |
| 50,000-100,000/μL | Increased post-traumatic bleeding risk |
| 20,000-50,000/μL | Significant risk with trauma or procedures |
| <20,000/μL | Spontaneous bleeding possible |
| <5,000-10,000/μL | Spontaneous bleeding likely; CNS hemorrhage risk |
- Robbins & Kumar Basic Pathology, p. 428; Goldman-Cecil Medicine, p. 1807
Causes of Thrombocytopenia
Three major mechanisms underlie all causes:
1. Decreased Platelet Production
Generalized Bone Marrow Failure:
- Aplastic anemia (congenital and acquired)
- Marrow infiltration: leukemia, disseminated cancer, myelophthisis
Selective Impairment of Platelet Production:
- Drug-induced: alcohol, thiazides, cytotoxic drugs
- Infections: measles, HIV infection
Ineffective Megakaryopoiesis:
- Megaloblastic anemia (B12/folate deficiency)
- Paroxysmal nocturnal hemoglobinuria (PNH)
2. Increased Platelet Destruction
A. Immunologic Destruction:
| Mechanism | Examples |
|---|---|
| Autoantibody (IgG vs. GP IIb/IIIa or Ib/IX) | Primary ITP, SLE, HIV |
| Alloantibody | Neonatal alloimmune thrombocytopenia, post-transfusion purpura |
| Drug-dependent antibody | Quinine, quinidine, sulfonamides |
| IgG-Fc/PF4-heparin complex | Heparin-induced thrombocytopenia (HIT) |
B. Non-Immunologic Destruction:
- Disseminated intravascular coagulation (DIC)
- Thrombotic thrombocytopenic purpura (TTP)
- Hemolytic uremic syndrome (HUS)
- Microangiopathic hemolytic anemias
- Cardiopulmonary bypass / intravascular catheters
- Septicemia / systemic inflammation
3. Splenic Sequestration
- Hypersplenism from portal hypertension, malignant hematologic disease, hereditary spherocytosis, sickle cell disease
- Normally ~one-third of platelets reside in the spleen; an enlarged spleen can sequester up to 90%
4. Dilutional
- Multiple transfusions for massive blood loss
- "Pseudothrombocytopenia" (in vitro EDTA-induced clumping - exclude first!)
- Henry's Clinical Diagnosis & Management by Laboratory Methods, p. 966; Goldman-Cecil Medicine, p. 1808
Key Clinical Syndromes
Immune Thrombocytopenic Purpura (ITP)
Pathogenesis: Autoantibodies (mainly IgG) target platelet membrane glycoproteins IIb/IIIa or Ib/IX. Antibody-coated platelets are destroyed in the spleen. The spleen is also the main site of antibody production. Bone marrow shows a compensatory increase in megakaryocytes.
Two forms:
- Acute ITP: Children 2-6 years, follows viral infection by ~3 weeks. Platelet count usually <20,000/mm³. Self-limited with >90% spontaneous remission.
- Chronic ITP: Women aged 20-40 years predominately. Insidious onset. Persistent beyond 12 months.
ITP Chronicity Classification (Rosen's EM):
- Newly diagnosed: <3 months
- Persistent: 3-12 months
- Chronic: >12 months
Clinical features of ITP:
- Petechiae, ecchymoses
- Easy bruising
- Epistaxis, gum bleeding
- Menorrhagia
- Hemorrhage after minor trauma
- Splenomegaly is NOT a feature of uncomplicated ITP
- Intracerebral hemorrhage is uncommon but dangerous
Heparin-Induced Thrombocytopenia (HIT)
Pathogenesis: IgG antibodies form against a platelet factor 4 (PF4)-heparin complex. These immune complexes bind platelet FcγRII receptors, causing platelet activation. The paradox: thrombocytopenia co-exists with a prothrombotic state.
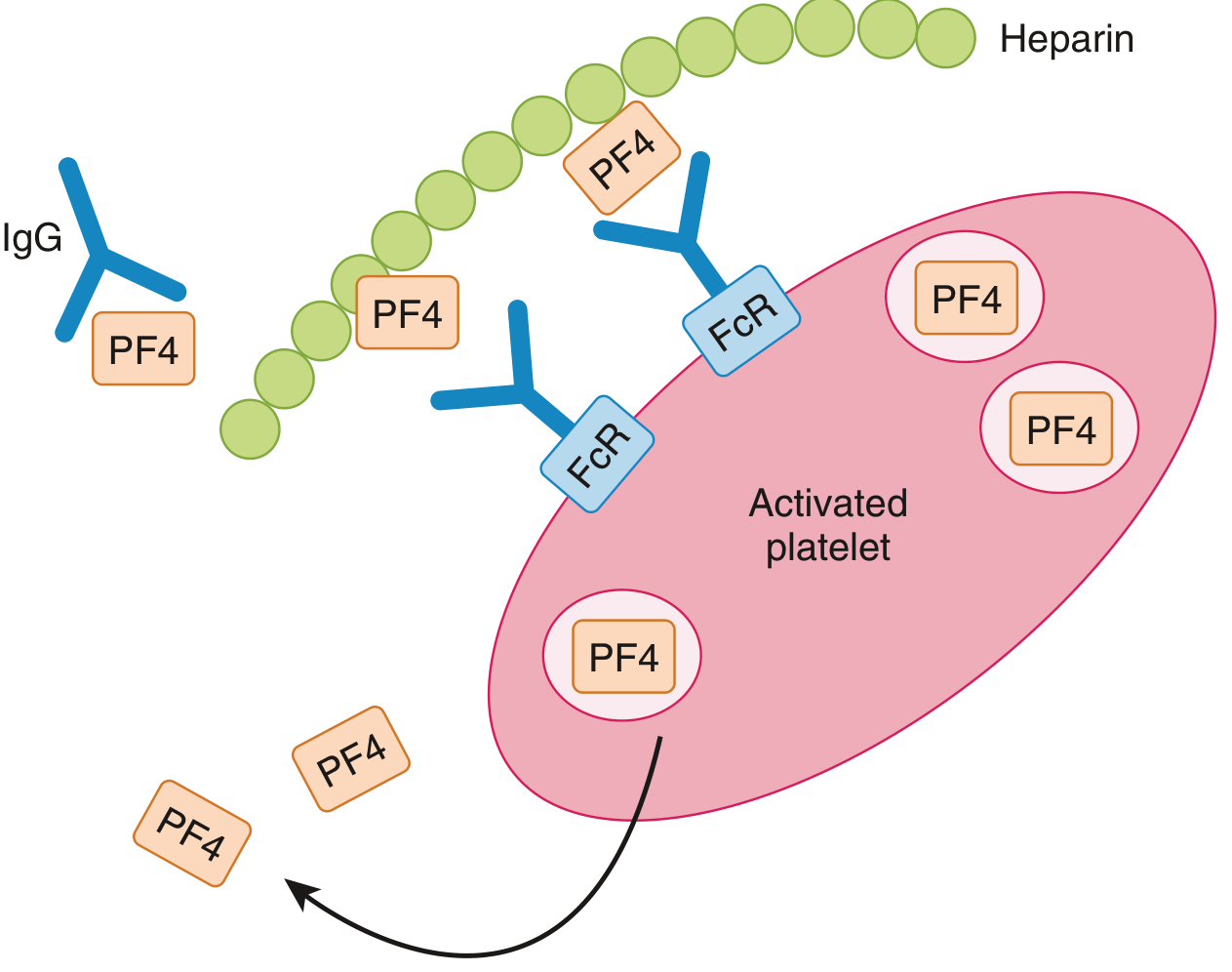
Figure: HIT mechanism - heparin-PF4-IgG complexes activate platelets via FcγRII receptors, perpetuating the cycle - Goldman-Cecil Medicine
Key features:
- Occurs in 3-5% of patients on unfractionated heparin (lower risk with LMWH)
- Onset 5-14 days after starting heparin (or rapidly on re-exposure)
- Both venous AND arterial thrombosis despite low platelet count
- Can cause severe morbidity (limb loss) and death
Thrombotic Thrombocytopenic Purpura (TTP)
Classic Pentad:
- Fever
- Thrombocytopenia
- Microangiopathic hemolytic anemia (MAHA)
- Transient neurologic deficits
- Renal failure
Pathogenesis: Congenital or acquired deficiency of the metalloprotease ADAMTS13, which normally cleaves ultra-large von Willebrand factor multimers. These accumulate and cause widespread platelet-rich microvascular thrombi.
Hemolytic Uremic Syndrome (HUS)
Shares MAHA + thrombocytopenia with TTP, but distinguished by:
- Predominant acute renal failure (not neurologic symptoms)
- Common in children
- Triggered by Shiga toxin (E. coli O157:H7) or abnormal complement activation
Clinical Features of Thrombocytopenia (General)
Bleeding pattern: Primarily mucocutaneous (small vessel) bleeding, NOT deep-tissue bleeding (which is more typical of coagulation factor deficiencies):
- Petechiae - 1-3 mm pinpoint non-blanching red spots (hallmark)
- Purpura - larger patches of skin hemorrhage
- Ecchymoses - bruising, often spontaneous
- Epistaxis (nosebleeds)
- Gingival bleeding
- Menorrhagia (heavy menstrual bleeding)
- GI bleeding - hematochezia, melena
- Hematuria
- CNS hemorrhage - most feared complication; occurs at counts <5,000-10,000/μL
Key distinction: Normal coagulation tests (PT, aPTT, fibrinogen) in isolated thrombocytopenia.
Management
General Principles
Platelet transfusion thresholds:
| Situation | Threshold |
|---|---|
| Spontaneous / prophylactic | <10,000/mm³ |
| Before central venous access | <20,000/mm³ |
| Before lumbar puncture | <50,000/mm³ |
| Before neurosurgery / ophthalmic surgery | <80,000-100,000/mm³ |
| Post-traumatic / active bleeding | <50,000/mm³ |
Note: Platelet transfusions are NOT useful in TTP, HIT, or ITP (unless life-threatening bleeding) - they may worsen thrombosis in TTP/HIT.
- Rosen's Emergency Medicine, p. 2470
ITP Management
First-line (newly diagnosed):
- Corticosteroids (prednisone 1 mg/kg/day or dexamethasone 40 mg/day x 4 days) - standard initial therapy
- IVIG (intravenous immunoglobulin) - rapid platelet rise; used for urgent situations
- Anti-D immunoglobulin - effective in Rh-positive non-splenectomized patients
Second-line (persistent/chronic):
- Splenectomy - durable response in 65-70% of patients; removes the main site of both antibody production and platelet destruction. Risk: lifelong susceptibility to encapsulated organisms.
- Rituximab - 375 mg/m² IV weekly x 4 doses; response ~60% but usually not durable (months). Risk: hepatitis B reactivation, PML.
Thrombopoietin Receptor Agonists (TPO-RAs):
- Eltrombopag (oral, daily) and avatrombopag (oral, daily)
- Romiplostim (SC, weekly)
- Stimulate megakaryocyte production; effect starts in 2-3 weeks; effective even post-splenectomy
- Can sustain long-term remission; risks: bone marrow fibrosis, thromboembolism
Third-line / Refractory ITP:
- Fostamatinib (Syk kinase inhibitor) - 100 mg twice daily; ~40-45% response rate
- Immunosuppressives: cyclophosphamide, azathioprine, cyclosporine, mycophenolate mofetil, dapsone
Emergency ITP (platelet <5,000/μL or internal bleeding):
- High-dose pulse corticosteroids + IVIG simultaneously
- Platelet transfusions may be given concurrently with IVIG for critical bleeding
- Goldman-Cecil Medicine, pp. 1097-1128
HIT Management
- Stop all heparin immediately (including LMWH, heparin flushes, heparin-coated catheters)
- Start a non-heparin anticoagulant: argatroban, bivalirudin, or fondaparinux
- Do NOT give platelet transfusions (worsens thrombosis risk)
- Do NOT start warfarin until platelets recover (risk of venous limb gangrene from protein C depletion)
TTP Management
- Plasma exchange (plasmapheresis) - primary treatment; replaces deficient ADAMTS13 and removes inhibitor antibodies. Life-saving.
- Corticosteroids - adjunctive
- Caplacizumab - anti-von Willebrand factor nanobody (newer agent); reduces time to platelet recovery
- Rituximab - used for immune-mediated (acquired) TTP
- Platelet transfusions are contraindicated (worsen microvascular thrombosis)
Drug-Induced Thrombocytopenia
- Stop the offending drug - platelet count typically recovers within days to a week
- Platelet transfusion if count <10,000-15,000/μL or life-threatening bleeding
- Common culprits: quinine/quinidine, sulfonamides, vancomycin, valproate, abciximab, tirofiban, furosemide, cimetidine, oxaliplatin
Decreased Production (Bone Marrow Causes)
- Treat underlying cause (stop myelosuppressive drugs, treat infection)
- Platelet transfusions are appropriate here (primary bone marrow disorder)
- For aplastic anemia: immunosuppression (ATG + cyclosporine) or stem cell transplantation
Diagnostic Approach Summary
- Check the blood smear first - exclude pseudothrombocytopenia (EDTA clumping artifact)
- CBC + differential - pancytopenia suggests bone marrow failure
- Review medications - drug-induced?
- PT/aPTT/fibrinogen/D-dimer - if abnormal, consider DIC
- Peripheral smear for schistocytes - fragmented RBCs = TTP/HUS/DIC
- Bone marrow biopsy - if decreased production suspected (increased megakaryocytes = peripheral destruction; decreased = production failure)
- 4T score for HIT assessment + anti-PF4/heparin antibodies
Recent Evidence: A 2025 network meta-analysis (PMID 40547032) found rhTPO, romiplostim, and eltrombopag all effective in pediatric ITP, with eltrombopag showing favorable efficacy-safety balance. A 2025 meta-analysis (PMID 38155484) confirmed the role of TPO-RAs in chemotherapy-induced thrombocytopenia.
Sources: Robbins & Kumar Basic Pathology (9780323790185); Goldman-Cecil Medicine (9780323930345); Henry's Clinical Diagnosis and Management (9780323673204); Rosen's Emergency Medicine (9780323757898)
This is a shared conversation. Sign in to Orris to start your own chat.