Glomerunephritis, definition, etiology, pathophysiology, pathogenesis, clinical feature, investigations, management
Reading File
Finding Sources
Searching PubMed
glomerulonephritis management
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
I now have all the information needed to compile a comprehensive answer. Let me write the full response.
Glomerulonephritis (GN)
1. Definition
Glomerulonephritis is a term encompassing a group of diseases characterized by inflammation of the glomeruli, typically immune-mediated, resulting in damage to the glomerular capillary walls. This leads to impaired filtration, leakage of blood and protein into the urine, and variable reduction in GFR. The hallmark clinical presentation is the nephritic syndrome: hematuria (with RBC casts), proteinuria, oliguria, azotemia, and hypertension.
Glomerular diseases presenting with a nephritic syndrome are characterized by inflammation in the glomeruli which damages capillary walls, causing red cell leakage (hematuria) and decreased GFR resulting in oliguria and azotemia. Hypertension results from both fluid retention and renin release from ischemic kidneys.
— Robbins & Kumar Basic Pathology, p. 510
2. Etiology
GN can be primary (idiopathic, confined to the kidney) or secondary (part of a systemic disease).
Primary GN
| Type | Key Feature |
|---|---|
| Postinfectious GN | β-hemolytic streptococcus (most common), Staphylococcus, Pneumococcus; also viral (mumps, measles, Hep B/C) |
| IgA Nephropathy | Most common primary GN worldwide; IgA deposits in mesangium |
| Membranoproliferative GN (MPGN) | Complement dysregulation or immune complex disease |
| Rapidly Progressive (Crescentic) GN | Anti-GBM antibodies, immune complexes, or pauci-immune (ANCA) |
| Minimal Change Disease | Cytokine-mediated podocyte injury |
Secondary GN
- Systemic lupus erythematosus (lupus nephritis)
- Henoch-Schönlein Purpura (IgA vasculitis)
- Goodpasture Syndrome (anti-GBM antibodies + pulmonary hemorrhage)
- ANCA-associated vasculitis (granulomatosis with polyangiitis, microscopic polyangiitis)
- Infective endocarditis, hepatitis B/C, HIV
- Diabetic nephropathy, amyloidosis
3. Pathogenesis
Three major immune mechanisms are recognized:
A. In Situ Immune Complex Formation
Antibodies bind directly to antigens planted within the glomerulus (e.g., streptococcal SpeB antigen in poststreptococcal GN, PLA2R antigen in membranous nephropathy). This is currently considered the predominant mechanism in poststreptococcal GN — exogenously planted antigens in subendothelial locations elicit inflammatory responses.
B. Circulating Immune Complex Deposition
Pre-formed antigen–antibody complexes from the circulation deposit in the GBM or mesangium. Classic in serum sickness, SLE, and some postinfectious GN. This mechanism is suggested by:
- Latent period compatible with time for antibody production
- Elevated anti-streptococcal antibody titers (ASO, anti-DNase)
- Low serum C3/CH50 (complement consumption)
- Granular IgG + C3 deposits on IF microscopy
C. Anti-GBM Antibody Disease
Autoantibodies target α3(IV) NC1 domain of type IV collagen in the GBM → Goodpasture disease. IF shows linear IgG staining along GBM.
D. Pauci-Immune (ANCA-Mediated)
No immune complex deposition detectable. ANCA (anti-PR3 or anti-MPO) activate neutrophils → endothelial damage → focal segmental necrotizing glomerulitis and crescents.
Downstream Mediators of Injury
Once immune complexes form, the following amplify injury:
- Complement activation (C3a, C5a → neutrophil/macrophage recruitment; MAC → direct cell lysis)
- Neutrophils and macrophages → release proteases, ROS, eicosanoids
- Platelets → aggregation at injury sites
- Cytokines: IL-1, TNF-α → endothelial activation; MCP-1 → monocyte chemotaxis; PDGF → mesangial proliferation; TGF-β → ECM deposition and sclerosis
- Coagulation cascade: fibrin deposition in Bowman's space → crescent formation (thrombin is a stimulus for crescent formation)
- Podocyte injury: foot process effacement, loss of slit diaphragm components (nephrin, podocin) → proteinuria
4. Pathophysiology
Acute Glomerular Injury
- Hypercellularity: proliferation of mesangial and endothelial cells + infiltration of neutrophils/monocytes → narrows capillary lumens → ↓ GFR
- Capillary wall disruption: allows RBC leakage → hematuria and RBC casts
- ↓ GFR → sodium and water retention → edema and hypertension
- Proteinuria: usually sub-nephrotic in most GN forms (except membranous or RPGN)
Crescent Formation (in RPGN)
Severe capillary injury → rupture of GBM → coagulation factors and inflammatory cells enter Bowman's space → parietal epithelial cell proliferation + monocyte/macrophage migration → crescents that compress the glomerular tuft → rapid loss of nephrons.
Chronic Progression
Once GFR falls to ~30–50% of normal, a self-sustaining cycle of glomerulosclerosis ensues regardless of initial cause:
- Hyperfiltration in remaining nephrons → increased capillary pressure
- TGF-β, PDGF → mesangial matrix expansion and glomerulosclerosis
- Proteinuria itself is tubulotoxic → tubular atrophy and interstitial fibrosis
5. Pathology (Morphology)
Postinfectious GN
- Light microscopy: Diffuse endocapillary hypercellularity (proliferation of endothelial and mesangial cells + neutrophil infiltration); affects nearly all glomeruli
- Immunofluorescence: Granular deposits of IgG and C3 in capillary walls and mesangium ("starry sky" pattern)
- Electron microscopy: Subepithelial "humps" (electron-dense deposits nestled against GBM)
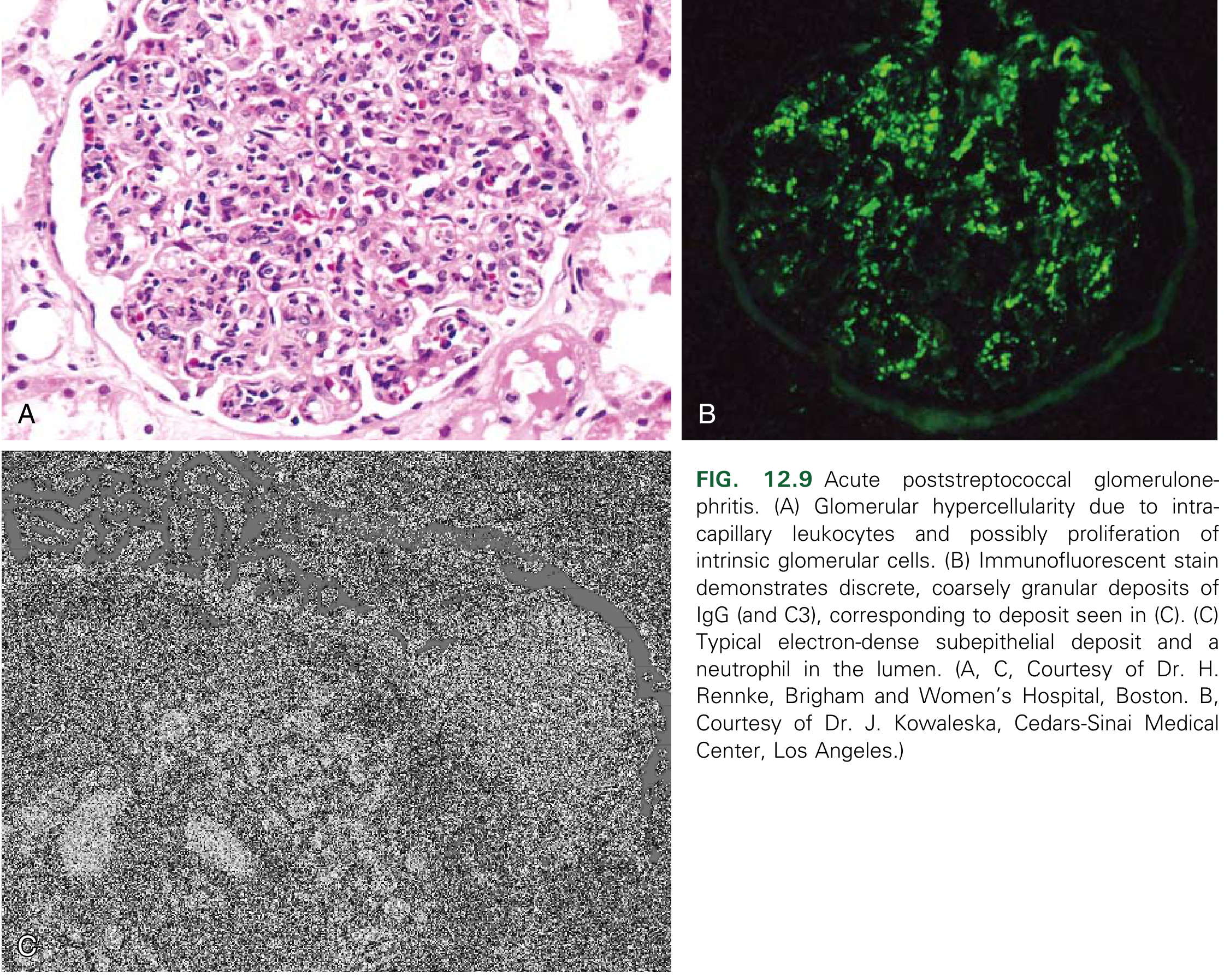
Rapidly Progressive (Crescentic) GN
- Light microscopy: Cellular crescents in Bowman's space (proliferating parietal epithelial cells + monocytes/macrophages + fibrin strands); segmental fibrinoid necrosis of capillary loops
- IF: Linear IgG (anti-GBM type), granular Ig/C3 (immune complex type), or negative (pauci-immune)
- EM: GBM ruptures; immune complex deposits in immune complex–mediated cases
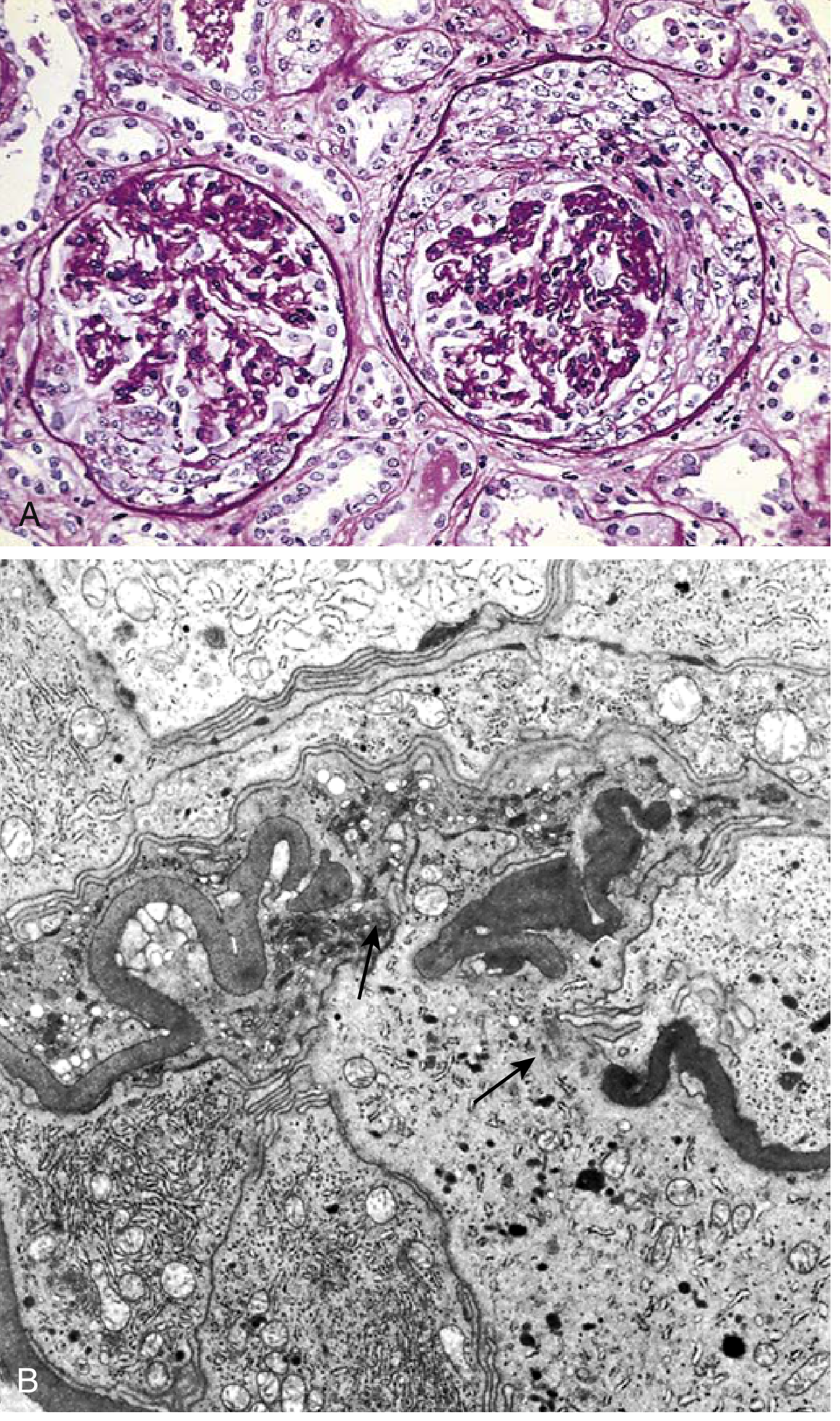
6. Classification
| Type | Histologic Pattern | IF Pattern | EM Deposits |
|---|---|---|---|
| Poststreptococcal GN | Diffuse endocapillary proliferative | Granular IgG + C3 | Subepithelial humps |
| IgA Nephropathy | Mesangial proliferative | IgA dominant, mesangial | Mesangial dense deposits |
| Membranoproliferative GN | Lobular proliferation + double contours | Granular C3 ± Ig | Subendothelial deposits |
| Anti-GBM GN | Crescentic ± diffuse proliferative | Linear IgG + C3 | No deposits |
| Pauci-immune (ANCA) GN | Necrotizing crescentic | Negative/scanty | No deposits |
| Lupus Nephritis | Variable (class I–VI) | "Full house" (IgG, IgM, IgA, C3, C1q) | Multiple locations |
7. Clinical Features
Nephritic Syndrome (classic presentation)
- Hematuria: macroscopic ("cola-colored" or "smoky" urine) or microscopic; RBC casts are pathognomonic
- Proteinuria: typically 1–3 g/day (sub-nephrotic); may reach nephrotic range in severe cases
- Oliguria: reduced urine output
- Azotemia: elevated BUN and creatinine
- Hypertension: often prominent, due to fluid retention + renin-angiotensin activation
- Edema: periorbital (facial), lower extremity
Systemic symptoms (50% of cases)
- Headache, malaise, anorexia
- Flank pain (due to renal capsule swelling)
Type-Specific Features
| Type | Distinguishing Features |
|---|---|
| Poststreptococcal GN | History of pharyngitis (1–3 wk) or skin infection (2–6 wk) before nephritis; more common in children 6–10 yr |
| RPGN | Rapid deterioration to acute renal failure within weeks; pronounced oliguria/anuria |
| Goodpasture Syndrome | Hemoptysis + hematuria (pulmonary-renal syndrome) |
| ANCA vasculitis | Sinusitis, nasal crusting, saddle nose deformity (GPA); systemic vasculitis features |
| IgA Nephropathy | Synpharyngitic hematuria (within 1–2 days of URTI) |
| Lupus Nephritis | Systemic SLE features (butterfly rash, arthritis, serositis) |
8. Investigations
Urine Analysis
- Urinalysis: Hematuria, proteinuria, granular casts, RBC casts (most specific for GN)
- Urine protein:creatinine ratio or 24-hr urine protein
- Urine microscopy: dysmorphic RBCs (acanthocytes)
Blood Tests
| Test | Finding | Significance |
|---|---|---|
| BUN, Creatinine | Elevated | Degree of azotemia |
| eGFR | Reduced | Severity of renal impairment |
| Serum C3, C4, CH50 | Low C3 ± normal C4 (alternate pathway); Low C3 + C4 (classical pathway) | Complement consumption pattern |
| ASO titre, anti-DNase B, Streptozyme | Elevated (positive in 80–95%) | Poststreptococcal GN |
| ANA, anti-dsDNA, anti-Smith | Positive | Lupus nephritis |
| ANCA (PR3-ANCA, MPO-ANCA) | Positive | GPA, MPA, pauci-immune crescentic GN |
| Anti-GBM antibody | Positive | Goodpasture disease |
| Serum IgA | Elevated in ~50% | IgA nephropathy |
| Cryoglobulins, HCV/HBV serology | Positive | Viral-associated GN |
| CBC | Anemia, leukocytosis | Inflammation, azotemia |
| Throat/skin culture | Group A Streptococcus | Confirm PSGN |
Imaging
- Renal ultrasound: Assesses kidney size; large/normal kidneys suggest acute process; small kidneys suggest chronic GN
- Chest X-ray: Pulmonary infiltrates/hemorrhage in Goodpasture/ANCA vasculitis
Renal Biopsy (Definitive)
Indicated when the diagnosis is uncertain or specific therapy is being considered. Evaluated by:
- Light microscopy (H&E, PAS, silver stain, Masson's trichrome)
- Immunofluorescence (Ig classes, complement)
- Electron microscopy (location of deposits)
In poststreptococcal GN, a biopsy is rarely required as the diagnosis can usually be made clinically and serologically.
9. Management
A. Postinfectious (Poststreptococcal) GN
Treatment is supportive — there is no role for immunosuppressive therapy, even when crescents are present:
- Antihypertensives (loop diuretics for volume-related hypertension; calcium channel blockers or ACEI)
- Diuretics (loop diuretics for edema and fluid overload)
- Dietary restriction (salt and fluid restriction, protein restriction if severe azotemia)
- Dialysis if severe AKI
- Antibiotics for active streptococcal infection (penicillin; does not prevent nephritis but prevents spread to contacts)
- Prognosis: Excellent in children — >95% complete recovery; 3–10% may have persistent hematuria/proteinuria; worse in adults
B. Rapidly Progressive (Crescentic) GN
Aggressive treatment is needed given rapid progression to ESRD:
- High-dose IV methylprednisolone (pulse steroids) followed by oral prednisolone
- Cyclophosphamide (oral or IV) — the standard cytotoxic agent
- Plasmapheresis (plasma exchange):
- Mandatory in anti-GBM disease (Goodpasture): removes circulating anti-GBM antibodies; can reverse pulmonary hemorrhage and renal failure
- Also beneficial in ANCA-associated crescentic GN
- Rituximab: increasingly used as an alternative to cyclophosphamide in ANCA-vasculitis
- Prognosis correlates with fraction of crescents: patients with <80% crescents have better outcomes
- Despite therapy, many patients require chronic dialysis or transplantation if diagnosed late
C. ANCA-Associated GN (GPA/MPA)
- Induction: cyclophosphamide + high-dose glucocorticoids OR rituximab + glucocorticoids
- Maintenance: azathioprine, mycophenolate mofetil, or rituximab
- Plasmapheresis for patients with severe AKI or pulmonary hemorrhage
D. Lupus Nephritis
- Class III/IV (proliferative): mycophenolate mofetil or cyclophosphamide + corticosteroids (induction); MMF or azathioprine (maintenance)
- Belimumab (anti-BLyS) and voclosporin are newer approved agents
E. IgA Nephropathy
- ACEI/ARBs to reduce proteinuria and control BP
- Corticosteroids for persistent proteinuria >1 g/day
- Supportive: low-sodium diet, treat infections promptly
F. General Supportive Measures (All GN)
- Blood pressure control (target <125/75 mmHg in proteinuric GN)
- ACEI/ARBs — reduce intraglomerular pressure and proteinuria (renoprotective)
- Fluid and salt restriction
- Dietary protein restriction in advanced AKI
- Treat hyperlipidemia
- Renal replacement therapy (dialysis/transplantation) for ESRD
Sources
- Robbins & Kumar Basic Pathology (Robbins Pathology), Chapter 12 — Kidney; pp. 509–511
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 20 — Kidney; pp. 835–843
- Harrison's Principles of Internal Medicine, 22E (2025), Chapter 375 — pp. 2460–2461
- Comprehensive Clinical Nephrology, 7th Edition, Chapter 25
- Brenner and Rector's The Kidney, 2-Volume Set
This is a shared conversation. Sign in to Orris to start your own chat.