A 30-year-old man develops weakness of all four limbs over 2 days; it began suddenly, was ascending and completed within 24 hours. There was fever and loose stools 2 weeks earlier; examination shows areflexic quadriparesis without sensory loss or bowel/bladder involvement. 1. a) What is your diagnosis? 2. b) Discuss the differential diagnosis and the laboratory & imaging features. 3. c) How will you manage the case?
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have all the content needed. Let me compose the comprehensive answer.
Clinical Case Analysis: Acute Flaccid Quadriparesis
a) Diagnosis
Guillain-Barré Syndrome (GBS) - Acute Inflammatory Demyelinating Polyradiculoneuropathy (AIDP)
This is a classic presentation:
- 30-year-old male
- Antecedent illness (fever + loose stools) 2 weeks prior - the typical post-infectious trigger
- Ascending weakness completed within 24 hours - rapid progression
- Areflexic quadriparesis - the invariable hallmark
- No sensory loss (pure motor), no bowel/bladder involvement - consistent with the motor-predominant form
The preceding gastroenteritis 2 weeks earlier is the most important clue - Campylobacter jejuni is the single most commonly identified precipitant, occurring in 25-50% of GBS cases, particularly preceding the axonal subtypes.
b) Differential Diagnosis and Laboratory / Imaging Features
GBS in Context
GBS has become the leading cause of acute paralytic disease in the Western world since the eradication of polio, with a mean annual incidence of 1.8 per 100,000 population. - Bradley & Daroff's Neurology in Clinical Practice
Required features for diagnosis (Asbury & Cornblath criteria):
- Progressive weakness of both legs and arms
- Areflexia or hyporeflexia
Supportive clinical features:
- Progression over days to 4 weeks
- Relative symmetry
- Mild sensory symptoms/signs
- Bifacial palsies
- Autonomic dysfunction
- Absence of fever at onset
- Recovery beginning 2-4 weeks after progression ceases
GBS Subtypes (Box 106.11, Bradley & Daroff)
| Subtype | Key Features |
|---|---|
| AIDP (most common in Europe/North America) | Demyelinating; post-infectious; sensory + motor |
| AMAN (acute motor axonal neuropathy) | Pure motor; axonal; summer epidemics in China; post-C. jejuni |
| AMSAN | Motor + sensory axonal; severe; poor recovery |
| Miller-Fisher syndrome | Ophthalmoplegia, ataxia, areflexia; anti-GQ1b antibody |
| Pharyngeal-cervical-brachial variant | Cranial nerve palsies |
| Acute pandysautonomia | Autonomic failure without somatic involvement |
In this case - pure motor, post-diarrheal illness - AMAN is a strong contender alongside AIDP.
Differential Diagnosis
Neuromuscular junction disorders:
- Myasthenia gravis (myasthenic crisis): fatigable weakness, ptosis, diplopia; CMAP decrement on slow repetitive stimulation; anti-AChR antibodies
- Botulism: descending paralysis, diplopia, dry mouth; contaminated food; presynaptic defect; incremental response on high-frequency stimulation; normal reflexes early
- Tick paralysis: ascending flaccid paralysis identical to GBS; tick found on scalp; resolves on tick removal
Anterior horn cell:
- Acute poliomyelitis / West Nile virus / enterovirus myelitis: asymmetric, fever throughout; CSF shows pleocytosis; no sensory loss; irreversible deficit
Peripheral nerve / roots:
- Acute intermittent porphyria (AIP): ascending motor neuropathy; abdominal pain; urine turns dark; elevated urinary delta-ALA and porphobilinogen
- Diphtheritic polyneuropathy: palatal palsy first; history of pharyngitis; no prodromal diarrhea
- Critical illness polyneuropathy: axonal neuropathy in ICU patients with sepsis; normal CSF
- Vasculitic neuropathy: painful, asymmetric; mononeuritis multiplex; elevated ESR/CRP
- Heavy metal poisoning (arsenic, thallium): history of exposure; Mees' lines; elevated urine metals
Spinal cord disorders (MUST exclude urgently):
- Transverse myelitis: upper motor neuron signs develop; sensory level on trunk; bladder/bowel involvement early; CSF pleocytosis; MRI shows cord signal change
- Cord compression / epidural abscess: spinal tenderness; sensory level; MRI definitive
- Anterior spinal artery infarction: sudden onset; pain at onset; spares dorsal columns (vibration/proprioception preserved)
Metabolic:
- Hypokalemic periodic paralysis, severe hypophosphatemia, hypermagnesemia
The key distinguishing features for this case are summarized below:
| Feature | GBS | Transverse Myelitis | Polio | Botulism |
|---|---|---|---|---|
| Reflexes | Absent | Increased (later) | Absent | Normal early |
| Sensory level | Absent | Present | Absent | Absent |
| Bladder/bowel | Late/spared | Early, prominent | Absent | May occur |
| CSF | Albuminocytologic dissociation | Pleocytosis | Pleocytosis | Normal |
| MRI spine | Normal | Signal change | Normal | Normal |
- Bradley & Daroff's Neurology in Clinical Practice; Rosen's Emergency Medicine
Laboratory and Imaging Features of GBS
1. CSF Examination (Lumbar Puncture)
The hallmark is albuminocytologic dissociation:
- Elevated CSF protein (>45 mg/dL) - found in almost all patients after the first week
- Normal or near-normal cell count (<10 cells/µL)
- In the first week, CSF protein may still be normal in up to 50% of patients - so a normal early LP does not exclude GBS
- Transient oligoclonal IgG bands and elevated myelin basic protein may be detected
- Moderate pleocytosis (>50 cells) should raise suspicion for HIV or Lyme disease-associated GBS
2. Nerve Conduction Studies (NCS) / Electromyography (EDX)
The most specific confirmatory test. In AIDP:
- Reduced motor conduction velocities (<80% of lower limit of normal)
- Prolonged distal latencies
- Partial conduction block - hallmark of acquired demyelination
- Absent or prolonged F-wave latencies (reflecting proximal/root involvement)
- Normal or mildly reduced CMAP amplitudes (early)
- Sensory nerve action potentials (SNAPs) may be reduced or absent
In AMAN:
- Low-amplitude CMAPs with preserved or near-normal conduction velocities
- Absent SNAPs
Note: EDX may be normal in the first 1-2 weeks; serial studies are required.
3. Serological / Other Blood Tests
- Anti-ganglioside antibodies:
- Anti-GM1, anti-GD1a - associated with AMAN (post-Campylobacter)
- Anti-GQ1b - Miller Fisher syndrome (>98% sensitivity)
- Stool culture / serology for C. jejuni
- CBC, electrolytes, LFTs (mild transient LFT elevation in ~1/3 of patients)
- Urine porphyrins (to exclude AIP)
- HIV, Lyme serology if CSF shows pleocytosis
- Anti-AChR antibodies if MG suspected
- Heavy metals (arsenic, thallium) if exposure suspected
4. Imaging
- MRI of the spine: Perform urgently to exclude cord compression, transverse myelitis, or epidural abscess - the MRI in GBS is normal or may show gadolinium enhancement of the cauda equina/nerve roots (reflecting blood-nerve barrier disruption). This is a supportive finding, not diagnostic.
- MRI brain: Usually normal; rarely shows brainstem findings in Miller Fisher or overlap syndromes.
- Chest X-ray: To assess respiratory compromise and detect aspiration pneumonia.
5. Pulmonary Function Tests
- Forced vital capacity (FVC) - critical for monitoring
- Negative inspiratory force (NIF) - bedside assessment of diaphragmatic strength
- The "20-30-40 rule" for intubation: FVC <20 mL/kg, NIF <-30 cmH₂O, PaO₂ <40 (on air) signals impending respiratory failure
c) Management
This patient has areflexic quadriparesis - he is already severely affected and requires hospital admission, likely ICU-level monitoring. Management involves three tracks: supportive care, specific immunotherapy, and rehabilitation.
Management Flowchart (Bradley & Daroff's Neurology)
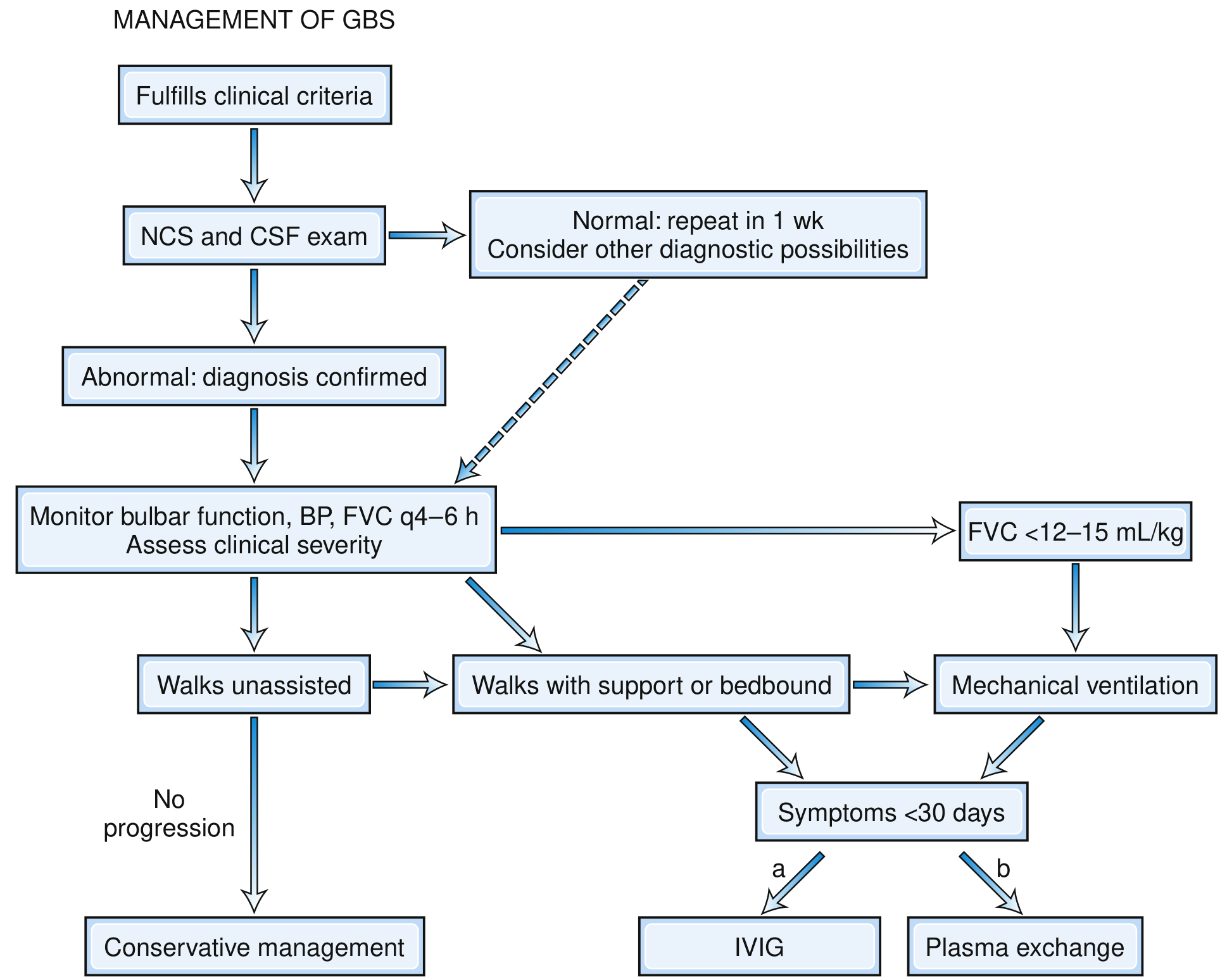
Track 1: Monitoring and Supportive Care
Respiratory:
- Measure FVC and NIF every 4-6 hours
- Indications for intubation and mechanical ventilation: FVC <12-15 mL/kg (or <1 L), NIF weaker than -20 to -25 cmH₂O, rising PaCO₂, hypoxia (PaO₂ <70 mmHg), inability to count to 20 on a single breath, or rapid clinical deterioration
- About 25-30% of hospitalized GBS patients require mechanical ventilation
- Tracheostomy if ventilation anticipated >2 weeks
Cardiovascular/Autonomic:
- Continuous cardiac monitoring (ECG telemetry)
- Antihypertensives must be short-acting and titratable (e.g., IV labetalol for hypertensive surges)
- Treat hypotension with IV saline and vasopressors cautiously - autonomic instability means rapid swings
- Avoid tracheal suctioning without atropine pretreatment (vagal spells can cause asystole)
DVT/PE Prevention:
- Subcutaneous heparin or LMWH + pneumatic compression stockings in all immobilized patients
Nutrition:
- Impaired swallowing → enteral tube feeding early
- High-caloric protein diet
Pain:
- NSAIDs for radicular/back pain
- Opioids for severe pain
Nursing and rehabilitation:
- Regular repositioning to prevent pressure sores
- Protect eyes in facial diplegia (artificial tears, tape eyelids shut at night)
- Pad ulnar and fibular nerve pressure points
- Physical therapy early - prevents contractures and venous stasis
- Establish a reliable communication method before intubation (letter/phrase board)
- Psychological support for patient and family
Infections:
- Monitor for aspiration pneumonia and UTI (affect ~50% of ICU patients with GBS)
- Prompt treatment with antibiotics
Track 2: Specific Immunotherapy
Both modalities are equally effective - supported by six large RCTs (>600 patients) and confirmed by multiple Cochrane reviews. Combination of both provides no additional benefit.
Option A: Intravenous Immunoglobulin (IVIG) - Preferred
- Dose: 0.4 g/kg/day for 5 days (total 2 g/kg)
- Timing: Start within 2 weeks of onset for maximum benefit; effective up to 4 weeks
- Mechanism: Neutralizes pathogenic antibodies, blocks Fc receptors, modulates complement
- Advantages: Ease of administration, no venous access requirements for apheresis
- Contraindications: IgA deficiency (risk of anaphylaxis), severe renal impairment, congestive cardiac failure (fluid load)
Option B: Therapeutic Plasma Exchange (Plasmapheresis)
- Schedule: 4-5 exchanges (40-50 mL/kg each) on alternate days over ~10-14 days
- Replacement fluid: Saline and albumin
- Timing: Most effective within 2 weeks; still beneficial up to 30 days from onset
- Even mildly affected patients benefit from 2 exchanges; 4 exchanges optimal for moderate-severe disease; 6 exchanges show no additional benefit (French Cooperative Group)
- Contraindications: Active sepsis/bacteremia, active bleeding, severe cardiovascular instability, recent MI
- Complications: Venous access problems, pneumothorax from central line insertion, catheter septicemia
Important note: Corticosteroids are NOT beneficial in GBS and are not recommended (multiple RCTs have shown no benefit and potential harm).
Track 3: Rehabilitation and Prognosis
- Recovery begins 2-4 weeks after progression stops (plateau phase lasts 2-4 weeks)
- Prognosis:
- 15% mild course, remain ambulatory, recover fully
- 70% complete recovery within 12 months; 82% by 24 months
- 20% have residual motor weakness at 1 year
- 2-5% mortality (from autonomic complications, respiratory failure, nosocomial infections)
- Up to 5% relapse
- Poor prognostic indicators: Age >60, preceding diarrheal illness (as in this patient), rapid progression to nadir in <7 days, ventilator dependence, low distal CMAP amplitudes, CMV infection, hyponatremia
- Physical therapy and rehabilitation are started as early as clinically feasible
Summary Table for this Patient
| Priority | Action |
|---|---|
| Immediate | Admit to HDU/ICU; cardiac monitoring; serial FVC/NIF every 4-6 h |
| Investigations | LP (CSF protein, cells), NCS/EMG, anti-GM1 antibodies, stool culture for Campylobacter, HIV/Lyme serology, spine MRI to exclude cord lesion |
| Therapy | IVIG 0.4 g/kg/day × 5 days (preferred) OR plasmapheresis 4 exchanges alternate days |
| Supportive | LMWH for DVT prophylaxis, enteral nutrition, NSAIDs/opioids for pain, chest physio |
| Monitor | Respiratory parameters q4-6h, BP, HR, autonomic signs, bulbar function |
| Rehab | Early physiotherapy once stable; psychological support |
Sources: Bradley & Daroff's Neurology in Clinical Practice; Adams & Victor's Principles of Neurology, 12th Ed.; Goldman-Cecil Medicine; Rosen's Emergency Medicine
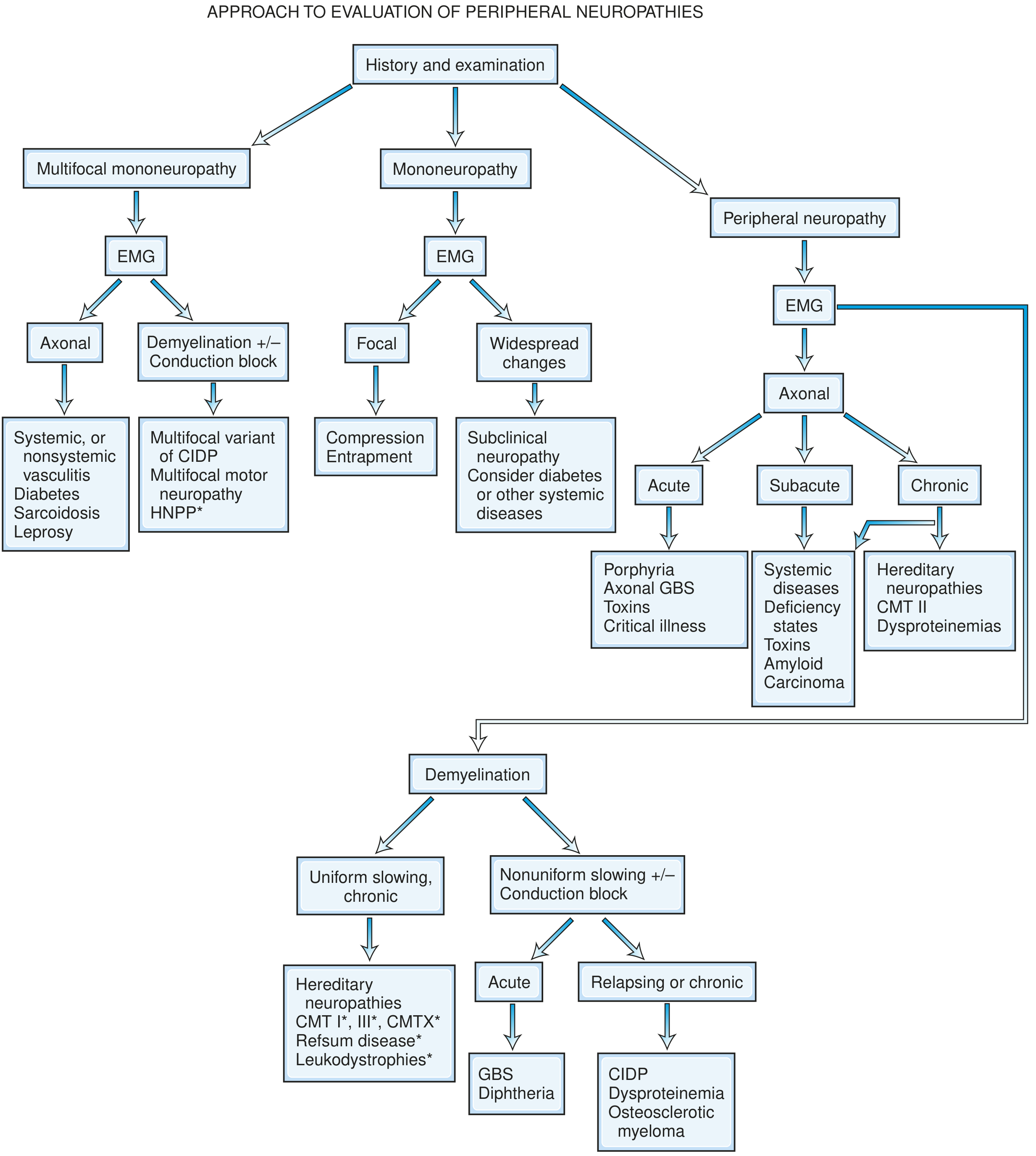
Diagnostic approach to peripheral neuropathy.
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
I now have all the material needed for a comprehensive, authoritative answer. Let me compile it.
Diagnostic Approach to Peripheral Neuropathy
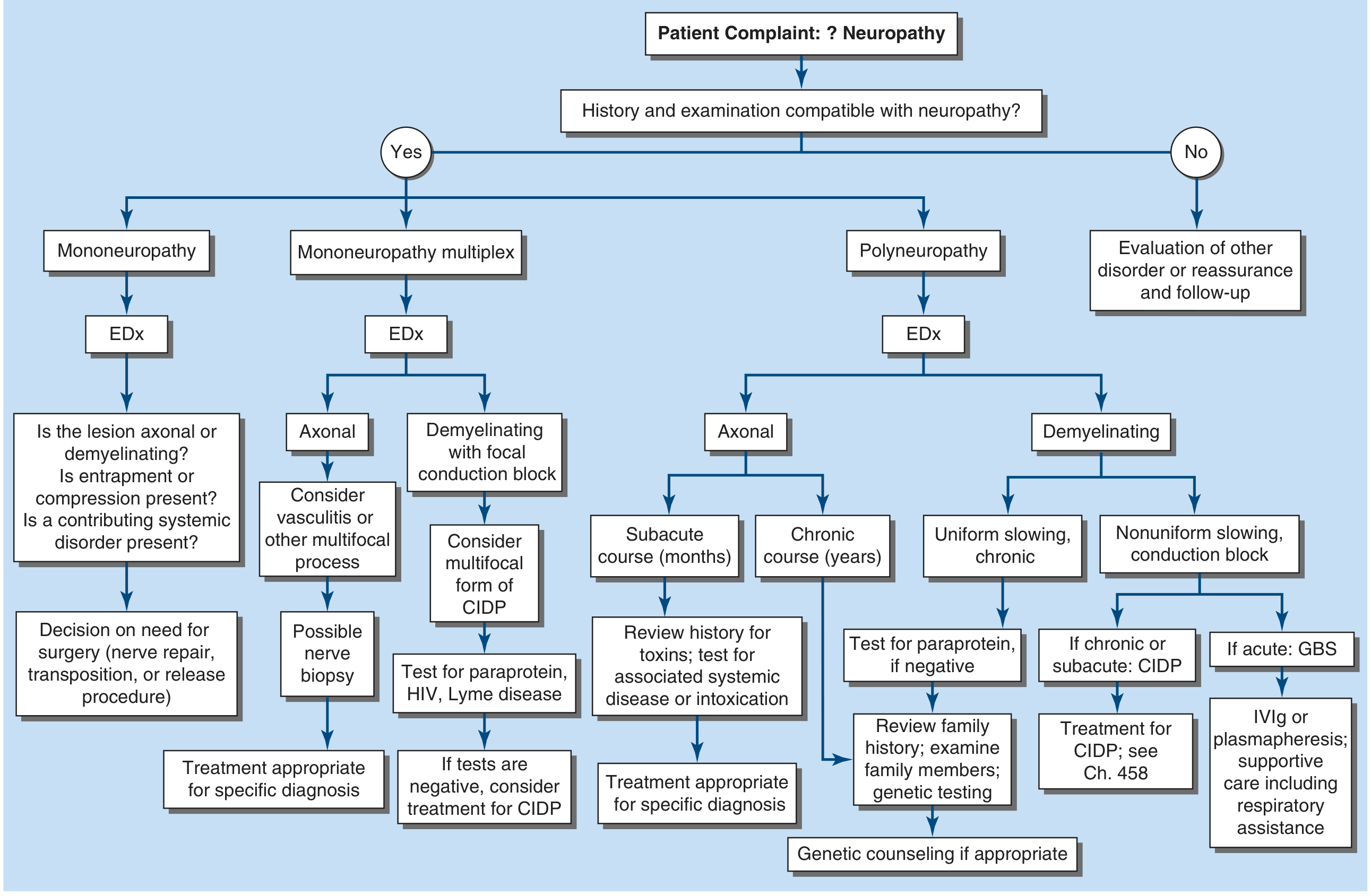
Peripheral neuropathy encompasses diseases of the peripheral nervous system (PNS) - the sensory, motor, and autonomic elements of peripheral nerves, their axons, myelin sheaths, and cell bodies. The diagnostic approach follows a logical sequence: establish the lesion is in the PNS → determine the topographic pattern → characterize the fiber type and pathology → determine the time course → identify the etiology. - Harrison's Principles of Internal Medicine, 22nd Ed.
Step 1: Confirm the Lesion is in the Peripheral Nervous System
The first task is to distinguish PNS disease from:
- Central nervous system (CNS) disorders - upper motor neuron signs (spasticity, hyperreflexia, extensor plantar response, sensory level), bladder/bowel involvement
- Neuromuscular junction (NMJ) disorders - fatigable weakness, no sensory involvement, normal reflexes
- Myopathies - proximal > distal weakness, normal sensation, elevated CK
Hallmarks of PNS disease:
- Weakness is flaccid with hyporeflexia or areflexia
- Sensory loss follows a nerve/root distribution or a "stocking-glove" pattern
- Fasciculations may be present (anterior horn cell or motor axon)
- No extensor plantar responses, no spasticity
Step 2: Determine the Topographic Pattern
At the outset, assign the patient to one of six syndromic patterns - Adams & Victor's Principles of Neurology, 12th Ed.:
| Pattern | Clinical Features | Key Clue |
|---|---|---|
| Polyneuropathy | Symmetric, length-dependent, distal > proximal | Stocking-glove loss; bilateral ankle areflexia |
| Radiculopathy / Polyradiculopathy | Root distribution; erratic asymmetric distribution; proximal + distal | Radicular pain; paraspinal denervation on EMG |
| Neuronopathy (motor or sensory) | Cell body affected; non-length-dependent | Sensory ataxia > weakness; asymmetric |
| Mononeuropathy | Single named nerve; focal | Entrapment, trauma, compression |
| Mononeuropathy multiplex | Multiple separate named nerves; asymmetric | Vasculitis, leprosy, diabetes |
| Plexopathy | Multiple nerves from one plexus | Post-radiation, neoplasm, trauma |
Step 3: The Seven Key Questions (Harrison's Framework)
Q1. What systems are involved?
Determine whether the neuropathy is:
- Motor only → motor neuropathy, NMJ disorder, myopathy; if asymmetric consider MND or multifocal motor neuropathy (MMN)
- Sensory only → most neuropathies are predominantly sensory; consider metabolic, toxic, hereditary
- Sensorimotor → most common pattern; acquired and hereditary
- Autonomic → especially amyloid, diabetes, GBS, porphyria; prominent autonomic dysfunction without diabetes should raise suspicion of amyloidosis
Q2. What is the distribution of weakness?
- Symmetric proximal + distal → acquired demyelinating (GBS, CIDP)
- Symmetric distal only → most acquired polyneuropathies (diabetes, toxic, metabolic)
- Asymmetric distal, multifocal → vasculitis, leprosy, mononeuropathy multiplex
- Asymmetric proximal + distal → polyradiculopathy, plexopathy
Q3. What is the nature of sensory involvement?
- Large fiber (vibration, proprioception loss, positive Romberg) → metabolic, CIDP, vitamin B12 deficiency
- Small fiber (pain, temperature loss; burning neuropathic pain; normal NCS) → diabetes/glucose intolerance, amyloid, idiopathic
- Both large and small fibers → most polyneuropathies
- Sensory ataxia predominant, non-length-dependent → sensory neuronopathy/ganglionopathy (Sjögren's, paraneoplastic anti-Hu, cisplatin)
Q4. Is there UMN involvement?
- UMN + peripheral neuropathy = combined system degeneration
- Most common cause: vitamin B12 deficiency (subacute combined degeneration)
- Others: adrenomyeloneuropathy, Friedreich's ataxia, HSP-plus, copper deficiency
Q5. What is the temporal evolution?
| Time Course | Likely Causes |
|---|---|
| Acute (days to 4 weeks) | GBS, porphyria, vasculitis, toxins, critical illness neuropathy |
| Subacute (4-8 weeks) | Inflammatory, toxic, paraneoplastic, deficiency states |
| Chronic (>8 weeks) | Hereditary CMT, CIDP, metabolic (diabetes), dysproteinemias |
| Relapsing-remitting | CIDP, porphyria, hereditary neuropathy with pressure palsies (HNPP) |
Q6. Is there a hereditary neuropathy?
Clues suggesting hereditary neuropathy (Bradley & Daroff):
- Family history of neuropathy
- Onset in childhood or adolescence
- Foot deformities (pes cavus, hammer toes) suggesting long-standing disease
- Lack of sensory symptoms despite sensory signs (sensory ataxia without pain)
- Uniform slowing on nerve conduction studies (CMT type 1: MCV <38 m/s)
- Consider genetic testing for CMT (PMP22 duplication/deletion, MPZ, GJB1/Cx32)
Q7. Are there associated medical conditions?
A comprehensive history must include:
- Diabetes mellitus, glucose intolerance
- Malignancy (paraneoplastic, direct infiltration, treatment-related)
- Connective tissue disease (Sjögren's, SLE, RA, vasculitis)
- Infections: HIV, Lyme, leprosy, hepatitis B/C, CMV, EBV
- Medications and toxins (see below)
- Alcohol use (direct toxicity + B1/B12 deficiency)
- Monoclonal gammopathy / plasma cell dyscrasia
- Preceding events (viral illness → GBS; vaccination)
Step 4: Fiber Type and Pathological Classification
By fiber type:
- Large myelinated fibers (Aα, Aβ): motor power, vibration, proprioception, deep tendon reflexes - assessed by NCS
- Small myelinated fibers (Aδ): pain, temperature, autonomic
- Unmyelinated fibers (C): pain, temperature, autonomic - assessed by skin biopsy IENFD, QSART
By pathology:
| Pathology | NCS Pattern | Examples |
|---|---|---|
| Axonopathy (dying-back) | Low CMAP/SNAP amplitude, mildly reduced velocity, active denervation on EMG | Diabetes, alcohol, toxins, uremia |
| Myelinopathy - acquired (demyelination) | Markedly reduced velocity, prolonged distal latency, conduction block, prolonged F-waves; non-uniform slowing | GBS (AIDP), CIDP, POEMS |
| Myelinopathy - hereditary | Uniform slowing (no conduction block) | CMT type 1, Refsum, leukodystrophies |
| Neuronopathy | Absent/low SNAPs with preserved motor; diffuse (non-length-dependent) | Paraneoplastic (anti-Hu), Sjögren's, cisplatin |
Step 5: Electrodiagnostic Studies (NCS / EMG)
NCS and EMG are the most important diagnostic tests and should be obtained early. They serve to:
- Confirm PNS pathology
- Determine axonal vs. demyelinating pathology
- Localize the lesion (root, plexus, nerve)
- Assess severity and prognosis
- Guide further workup
NCS parameters:
- CMAP amplitude - proportional to number of functioning motor axons
- Conduction velocity - reflects myelin integrity
- Distal latency - terminal segment conduction
- F-wave latency - proximal/root segment
- SNAP amplitude and latency - sensory axons
Needle EMG findings:
- Fibrillations and positive sharp waves - active denervation (axonal loss)
- Large polyphasic motor units - chronic reinnervation
- Reduced recruitment - motor unit loss
- Paraspinal fibrillations - polyradiculopathy
Diagnostic Flowcharts
Bradley & Daroff's approach - based on topography then EMG pathology:
Harrison's approach - starting from history/exam compatibility:
Step 6: Laboratory and Special Investigations
Tier 1 - All patients with generalized symmetric polyneuropathy (Harrison's):
| Test | Detects |
|---|---|
| HbA1c + fasting glucose | Diabetes (most common cause worldwide) |
| Oral glucose tolerance test | Prediabetes/impaired fasting glucose (in painful sensory neuropathy) |
| CBC, electrolytes, renal/hepatic function | Uremia, hepatic failure |
| Thyroid function (TSH) | Hypothyroid neuropathy |
| Vitamin B12 (± methylmalonic acid) | B12 deficiency / subacute combined degeneration |
| Serum protein electrophoresis + immunofixation | Monoclonal gammopathy (MGUS, myeloma, POEMS) |
| Free serum light chains (kappa/lambda ratio) | More sensitive for amyloid evaluation |
| ESR, ANA, rheumatoid factor | Connective tissue disease |
Tier 2 - Mononeuropathy multiplex / asymmetric / vasculitis suspected:
| Test | Detects |
|---|---|
| ANCA, cryoglobulins | Vasculitis, cryoglobulinemia |
| Hepatitis B + C serology | Hepatitis-associated neuropathy, cryoglobulinemia |
| HIV, Lyme (Western blot) | Infectious neuropathies |
| CMV titer | CMV-related neuropathy |
Tier 3 - Specific situations:
| Test | Indication |
|---|---|
| Anti-ganglioside antibodies (GM1, GQ1b, MAG) | GBS (AMAN), Miller Fisher, IgM paraprotein neuropathy |
| Anti-Hu, anti-Yo, anti-amphiphysin | Paraneoplastic sensory neuronopathy |
| Anti-SS-A/Ro, SS-B/La | Sjögren's-associated neuropathy |
| Urine porphyrins (delta-ALA, PBG) | Acute intermittent porphyria (axonal GBS pattern) |
| Heavy metals (arsenic, thallium, lead) in urine/blood | Toxic neuropathy (only if clinically suspected) |
| Hereditary neuropathy panel / genetic testing (PMP22, MPZ, GJB1) | Suspected CMT or hereditary neuropathy |
| Lumbar puncture / CSF | GBS, CIDP (albuminocytologic dissociation); HIV, Lyme, lymphoma |
| Skeletal survey (whole body X-ray) | Osteosclerotic myeloma in acquired demyelinating neuropathy with M-spike |
| Bone marrow biopsy | Monoclonal gammopathy → hematology referral |
Step 7: Special Tests for Specific Situations
Small-Fiber Neuropathy (normal NCS)
When NCS are normal but small-fiber neuropathy is suspected (burning pain, autonomic symptoms):
- Skin punch biopsy - intraepidermal nerve fiber density (IENFD) with anti-PGP 9.5 staining; loss of intraepidermal fibers confirms small-fiber neuropathy (diagnosis if ≥2 tests abnormal)
- Quantitative sensory testing (QST) - heat/cold pain thresholds
- Quantitative sudomotor axon reflex test (QSART) - 80% sensitivity for autonomic small-fiber involvement
Nerve Biopsy (sural or superficial peroneal sensory nerve)
Essential for diagnosis:
- Vasculitis (combined nerve + muscle biopsy increases yield from ~50% to 60-70%)
- Amyloidosis (light chain or hereditary)
- Sarcoidosis
- Leprosy / Hansen's disease
- Giant axonal neuropathy
- Tumor infiltration
Supportive (not essential):
- CMT type 1 and 3
- CIDP
- IgM paraprotein neuropathy (anti-MAG)
Neuromuscular Ultrasound
- Increasingly used; non-invasive
- Focal nerve enlargement (cross-sectional area) at entrapment sites: carpal tunnel, ulnar at elbow
- Multifocal enlargement at non-entrapment sites and brachial plexus trunks → highly specific for CIDP; distinguishes acquired from axonal/hereditary neuropathy
-
- Bradley & Daroff's Neurology in Clinical Practice
MRI
- MRI spine/nerve roots: Enhancement of cauda equina/roots in GBS, CIDP, lymphomatous radiculopathy
- MRI brachial plexus: Plexopathy (neoplastic, idiopathic, post-radiation)
- MRI brain: When central involvement or paraneoplastic syndrome suspected
Common Causes by Pattern - Practical Summary
| Pattern | Top Causes |
|---|---|
| Symmetric distal sensorimotor, axonal, chronic | Diabetes, alcohol, uremia, hypothyroid, B12 deficiency, toxins (drugs, heavy metals), CIDP |
| Symmetric proximal + distal, demyelinating, acute | GBS (AIDP) |
| Symmetric proximal + distal, demyelinating, chronic/relapsing | CIDP, dysproteinemia (POEMS, IgM-MAG), hereditary CMT I |
| Asymmetric, axonal, multifocal | Vasculitis, mononeuropathy multiplex, leprosy, sarcoid, Lyme |
| Pure motor, proximal + distal | GBS, CIDP, porphyria, lead toxicity, MMN |
| Small-fiber, normal NCS | Diabetes/prediabetes, amyloid, idiopathic |
| Sensory ataxia, non-length-dependent | Paraneoplastic (anti-Hu), Sjögren's, cisplatin, B6 toxicity |
| Autonomic predominant | Amyloid (FAP), diabetes, GBS, HSAN, porphyria |
Important note: Despite extensive evaluation, approximately 50% of patients with a predominantly sensory polyneuropathy have no etiology identified and are labeled cryptogenic sensory polyneuropathy (CSPN). - Harrison's Principles of Internal Medicine, 22nd Ed.
Skin, Hair, and Nail Clues on Examination
(Bradley & Daroff's):
| Finding | Disease |
|---|---|
| Purpura / livedo reticularis | Vasculitis, cryoglobulinemia |
| Angiokeratomas | Fabry disease |
| Skin hypopigmentation, anesthetic patches | Leprosy |
| Skin hyperpigmentation | POEMS syndrome |
| Alopecia | Thallium poisoning |
| Mees' lines (transverse white nail bands) | Arsenic / thallium toxicity |
| Ichthyosis | Refsum disease |
| Curly hair | Giant axonal neuropathy |
| Bullous skin lesions | Variegate porphyria |
Key Take-Home Points
- The history and examination alone narrow the differential dramatically - use the 7 key questions systematically.
- EDx (NCS + EMG) is the single most important test - it distinguishes axonal from demyelinating, localizes the lesion, and guides all further workup.
- HbA1c and glucose tolerance testing should be ordered in virtually all patients with distal sensory polyneuropathy - diabetes and prediabetes are the most common identifiable causes.
- Acute demyelinating neuropathy → GBS until proven otherwise; urgent CSF and hospital admission.
- Prominent autonomic dysfunction without diabetes → think amyloidosis.
- Non-length-dependent sensory ataxia → think sensory neuronopathy/ganglionopathy - screen for occult malignancy and Sjögren's.
- Uniform NCS slowing + family history + foot deformities → hereditary neuropathy (CMT) - genetic testing not biopsy.
- Conduction block on NCS → acquired demyelination; never seen in hereditary neuropathy.
Sources: Harrison's Principles of Internal Medicine 22nd Ed.; Bradley & Daroff's Neurology in Clinical Practice; Adams & Victor's Principles of Neurology 12th Ed.; Goldman-Cecil Medicine
This is a shared conversation. Sign in to Orris to start your own chat.