Biotramformation comcept do
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is a thorough, textbook-based explanation of Biotransformation:
Biotransformation
Definition
Biotransformation is the chemical modification of a drug (or other xenobiotic) within the body - primarily by the liver - that converts it into a different chemical form, usually more polar (water-soluble), so it can be excreted. It is the metabolic phase of pharmacokinetics (Absorption → Distribution → Biotransformation → Excretion).
"Biotransformation includes the chemical processes by which the drug molecule is altered in the body."
- Morgan and Mikhail's Clinical Anesthesiology, 7e
Sites of Biotransformation
| Site | Significance |
|---|---|
| Liver | Primary organ; contains microsomal enzymes (CYP450, UGT, GST, sulfotransferases) |
| Gastrointestinal tract | Intestinal wall contributes significantly (e.g., midazolam ~50% intestinal) |
| Lungs, Skin, Kidneys, Brain | Minor roles |
| Plasma/Tissues | Ester hydrolysis (e.g., ester-type local anesthetics like succinylcholine) |
After oral administration, many drugs (isoproterenol, morphine, lidocaine) undergo extensive first-pass hepatic metabolism before reaching systemic circulation. - Katzung's Basic & Clinical Pharmacology, 16e
The Two Phases of Biotransformation
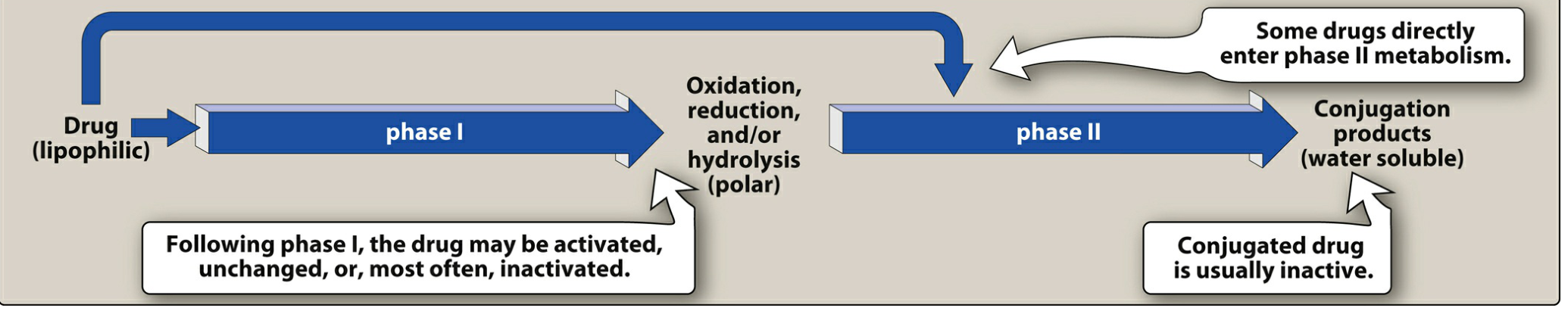
Phase I Reactions - "Functionalization"
Goal: Convert lipophilic parent drug into more polar metabolite by introducing or unmasking a functional group (-OH, -NH2, -SH).
Reactions:
- Oxidation (most common)
- Reduction
- Hydrolysis
Key enzyme system: Cytochrome P450 (CYP) - located in the smooth endoplasmic reticulum of hepatocytes. Requires NADPH and O2.
Effect on activity:
- Most often → inactivation of the drug
- Sometimes → activation (prodrugs, e.g., codeine → morphine)
- Sometimes → toxic metabolites (e.g., acetaminophen overdose → NAPQI via CYP2E1)
Non-CYP Phase I reactions:
- Amine oxidation (catecholamines, histamine)
- Alcohol dehydrogenation (ethanol)
- Esterase hydrolysis (aspirin)
Phase II Reactions - "Conjugation / Synthetic"
Goal: Couple a Phase I metabolite (or sometimes the parent drug directly) with an endogenous polar molecule to form a highly water-soluble conjugate excreted in urine or bile.
Conjugation substrates:
| Reaction | Endogenous Substrate | Key Enzyme | Examples |
|---|---|---|---|
| Glucuronidation | UDP-glucuronic acid | UDP-glucuronyl transferase (UGT) | Bilirubin, morphine, acetaminophen |
| Sulfation | PAPS (3'-phosphoadenosine-5'-phosphosulfate) | Sulfotransferase | Thyroxine, bile acids, acetaminophen |
| Acetylation | Acetyl-CoA | N-acetyltransferase (NAT) | Isoniazid, sulfonamides |
| Glutathione conjugation | Glutathione (GSH) | GSH S-transferase | Electrophilic metabolites (e.g., NAPQI) |
| Amino acid conjugation | Glycine, glutamine | - | Aspirin metabolites |
Result: Phase II products are usually pharmacologically inactive and excreted. A notable exception is morphine-6-glucuronide, which is more potent than morphine itself.
"Glucuronidation is the most common and the most important conjugation reaction." - Lippincott Pharmacology
Important Nuance: Phase II Before Phase I
The traditional sequence (Phase I → Phase II) is not absolute. For example:
- Isoniazid (INH): The hydrazide moiety first undergoes Phase II acetylation to form N-acetyl-INH, which then undergoes Phase I hydrolysis to isonicotinic acid. Phase II actually comes first here.
- Drugs with existing -OH, -NH2, or -COOH groups can bypass Phase I and go directly to Phase II conjugation.
Cytochrome P450 (CYP) System - Key Isoforms
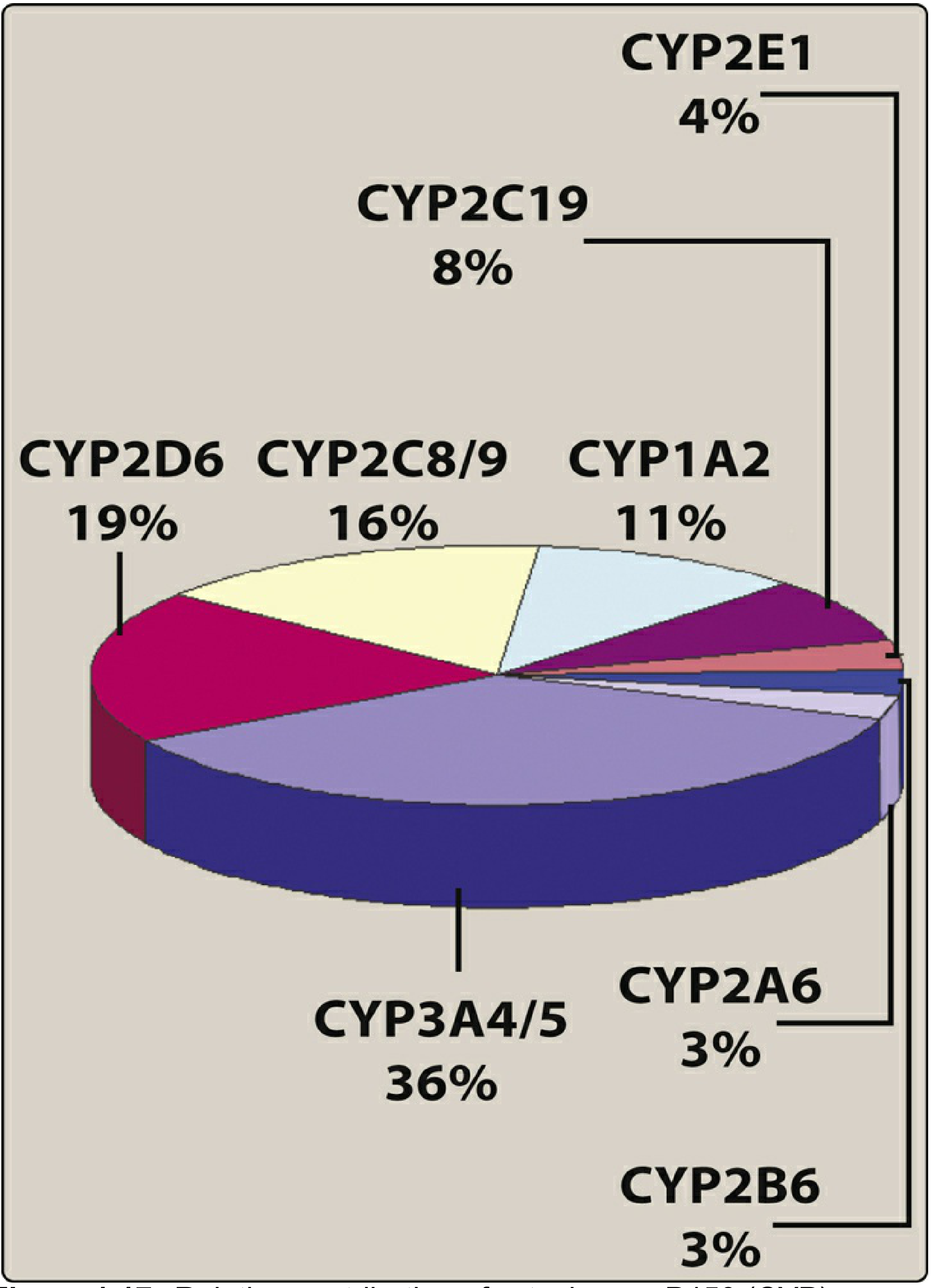
| Isoform | % Contribution | Key Substrates | Key Inducers | Key Inhibitors |
|---|---|---|---|---|
| CYP3A4/5 | 36% (largest) | Carbamazepine, cyclosporine, erythromycin, nifedipine, simvastatin | Rifampin, phenobarbital, phenytoin, carbamazepine | Ketoconazole, ritonavir, clarithromycin |
| CYP2D6 | 19% | Fluoxetine, haloperidol, paroxetine, propranolol | None significant | Fluoxetine, paroxetine |
| CYP2C8/9 | 16% | Warfarin, NSAIDs | Rifampin | Omeprazole |
| CYP1A2 | 11% | Theophylline, caffeine | Tobacco smoke, omeprazole | Fluvoxamine |
| CYP2C19 | 8% | PPIs, diazepam, clopidogrel | Rifampin | Omeprazole |
| CYP2E1 | 4% | Ethanol, acetaminophen | Ethanol, isoniazid | Disulfiram |
Hepatic Clearance Concepts
Extraction Ratio (ER) = fraction of drug removed by the liver in a single pass.
| Type | Extraction Ratio | Clearance depends on | Examples | Effect of enzyme induction |
|---|---|---|---|---|
| High ER (flow-dependent) | >0.7 | Liver blood flow | Propofol, lidocaine, morphine, nitroglycerin, propranolol | Minimal effect |
| Low ER (capacity-dependent) | <0.3 | Enzyme capacity | Methadone (10%), alfentanil (15%), warfarin | Significant increase in clearance |
Enzyme Induction and Inhibition
Induction = increased synthesis of CYP enzymes → faster drug metabolism → lower plasma levels → potential treatment failure.
- Classic inducers: Phenobarbital (broad, increases P450 reductase), rifampin, carbamazepine, tobacco smoke (induces CYP1A2)
Inhibition = competition or blocking of CYP → slower metabolism → drug accumulation → toxicity.
- Classic inhibitors: Ketoconazole, ritonavir, clarithromycin (inhibit multiple CYPs), omeprazole (warfarin interaction)
- Mechanisms: substrate competition, competitive inhibition, non-competitive inhibition, product inhibition
Factors Affecting Biotransformation Rate
- Age: Neonates and elderly have reduced CYP activity
- Genetics: Polymorphisms in CYP2D6, CYP2C19 → poor vs. extensive metabolizers
- Disease: Cirrhosis, hepatitis → reduced metabolic capacity
- Gender: Minor differences in CYP activity
- Drug-drug interactions: Induction/inhibition of shared CYP isoforms
- Diet: Grapefruit juice inhibits CYP3A4 in the gut wall
Biotransformation and Toxicity
Some drugs generate toxic reactive intermediates during Phase I metabolism:
- Acetaminophen: CYP2E1/CYP3A4 generates NAPQI (N-acetyl-p-benzoquinone imine) - normally detoxified by GSH. In overdose, GSH is depleted → hepatic necrosis.
- Isoniazid: CYP450 bioactivation → isonicotinic acyl radical → binds hepatic proteins → idiosyncratic immunotoxicity.
- Halothane, phenothiazines: Converted to reactive forms → cellular injury.
- Carcinogens: CYP450 enzymes activate aromatic carcinogens to their active mutagenic forms.
Sources:
- Morgan and Mikhail's Clinical Anesthesiology, 7e, pp. 265-266
- Lippincott Illustrated Reviews: Pharmacology, pp. 55-61
- Katzung's Basic and Clinical Pharmacology, 16e, pp. 93-95
- Mulholland and Greenfield's Surgery, 7e, pp. 92-94
- Tietz Textbook of Laboratory Medicine, 7e






+8 more
Make revesion flashcards of these topics from my book and mcqs for practise i will see Tommrow before starting micro and patho
analyse_media
analyse_media
analyse_media
analyse_media
analyse_media
Good luck tomorrow! Here are all your revision flashcards + MCQs based directly on your book pages:
📚 PHARMACOKINETICS - REVISION FLASHCARDS
TOPIC 1: BIOAVAILABILITY
CARD 1
Q: What is the FDA definition of bioavailability?
A: The rate at which AND the extent to which the active concentration of the drug is available at the desired site of action. Measured by AUC (area under concentration-time curve).
CARD 2
Q: What is the bioavailability via IV route? Why?
A: 100% - because drug is directly administered into systemic circulation.
CARD 3
Q: Why is oral bioavailability < 100%? (2 main reasons)
A:
- Incomplete absorption - drug not fully absorbed from GIT
- First-pass metabolism - drug metabolised in gut wall + liver before reaching systemic circulation
CARD 4
Q: Atenolol is too ________ → fails to cross lipid cell membrane. Acyclovir is too ________ → can't cross water layer.
A: Atenolol = too hydrophilic. Acyclovir = too lipophilic.
CARD 5
Q: What is the formula for Extraction Ratio (ER)?
A: ER = CL(liver) / Q
- CL(liver) = drug clearance by liver
- Q = hepatic blood flow (normally 90 L/hour in 70 kg adult)
CARD 6
Q: What is the formula for systemic bioavailability (F)?
A: F = f × (1 - ER)
Full form: F = f × (1 - CL(liver)/Q)
- f = fraction absorbed
- ER = extraction ratio
TOPIC 2: FACTORS AFFECTING BIOAVAILABILITY
CARD 7
Q: Order of bioavailability by formulation type (highest to lowest)?
A: Solution > Suspension > Tablet > Coated tablet > Capsule
CARD 8
Q: How does particle size affect bioavailability?
A: Smaller (microfine) particles = higher bioavailability due to better dissolution. Example: sodium tolbutamide > tolbutamide.
CARD 9
Q: Amorphous vs crystalline form - which has higher bioavailability?
A: Amorphous form has higher bioavailability (dissolves faster). Example: amorphous chloramphenicol palmitate > crystalline form.
CARD 10
Q: Which highly polar drugs are NOT absorbed orally (low bioavailability)?
A: Streptomycin, acetylcholine, neostigmine (too ionised/polar to be absorbed).
CARD 11
Q: How do chlorpromazine and propranolol affect each other's bioavailability when given together?
A: They compete for the same hepatic metabolising enzymes → reduced first-pass metabolism of each other → increased oral bioavailability of both.
CARD 12
Q: Effect of severe hepatic disease on oral bioavailability?
A: Decreases first-pass metabolism → increased oral bioavailability → risk of toxicity with usual doses.
CARD 13
Q: What is bioequivalence vs chemical equivalence?
A: Chemical equivalence = same amount of drug. Bioequivalence = same blood levels/pharmacological effect. Two formulations can be chemically equivalent but biologically inequivalent (e.g., different digoxin formulations).
CARD 14
Q: Classic example of bioinequivalence?
A: Phenytoin ('Dilantin sodium') in Australia 1968 - calcium sulphate replaced by lactose as diluent → lactose dissolved faster → toxic plasma levels of phenytoin.
TOPIC 3: VOLUME OF DISTRIBUTION (aVd)
CARD 15
Q: Define apparent Volume of Distribution (aVd).
A: The theoretical volume the body would need to have if the drug concentration throughout was the same as in plasma.
Formula: aVd = Total drug in body (Q) / Plasma concentration (Cp)
CARD 16
Q: Why is it called "apparent" Vd?
A: Because the body is not a single homogeneous compartment. It's a theoretical/calculated value.
CARD 17
Q: Drugs with HIGH plasma protein binding have ____ aVd. Example?
A: LOW aVd (drug stays in vascular compartment).
Example: Warfarin - 90% protein bound → aVd = 0.15 L/kg (≈ plasma volume).
CARD 18
Q: What are the consequences of HIGH plasma protein binding (PPB)? (4 points)
A:
- Low aVd (drug retained in vascular compartment)
- Acts as temporary storage → increases duration of action
- Dialysis has NO role in toxicity (can't filter protein-bound drug)
- Risk of displacement reactions when two highly bound drugs compete for same binding site
CARD 19
Q: Acidic drugs bind to ______. Basic drugs bind to ______.
A:
- Acidic drugs → Albumin (e.g., barbiturates, benzodiazepines, phenytoin, valproate, NSAIDs, penicillins, sulphonamides, tetracyclines)
- Basic drugs → α1-acid glycoprotein (e.g., verapamil, lignocaine, bupivacaine, quinidine, disopyramide, beta blockers)
CARD 20
Q: What 3 factors determine fraction of drug bound to plasma protein?
A:
- Number of binding sites
- Affinity of drug for plasma proteins
- Drug concentration
TOPIC 4: DRUG METABOLISM / BIOTRANSFORMATION
CARD 21
Q: Define biotransformation.
A: The process of converting lipid-soluble, non-polar (unionised) drugs into lipid-insoluble, ionised forms in the body to make them excretable by the kidney.
CARD 22
Q: Principal organ of drug metabolism? Other sites?
A: Liver is principal. Others: intestines (lumen + wall), lungs, skin, kidneys.
CARD 23
Q: What is first-pass metabolism / pre-systemic metabolism?
A: Metabolism that occurs when the drug passes through the gut wall + portal blood + liver for the first time, before reaching systemic circulation. Results in reduced bioavailability.
CARD 24
Q: Name drugs with VERY HIGH first-pass metabolism (not used orally)?
A: Testosterone, Morphine, Isoprenaline, Lignocaine, Hydrocortisone
Mnemonic: "The Man Is Lying Here"
CARD 25
Q: Name drugs with HIGH first-pass metabolism (low oral bioavailability)?
A: Propranolol, Alprenolol, Verapamil, Salbutamol, Nitroglycerine, Methyltestosterone, Pethidine
CARD 26
Q: What is Phase I reaction? What functional groups are introduced/unmasked?
A: Non-synthetic reactions. Introduce or unmask -OH, -SH, or -NH2 groups.
Types: Oxidation, Reduction, Hydrolysis.
Makes drug more polar/excretable. Metabolites may be inactive, active, or toxic.
CARD 27
Q: What is Phase II reaction?
A: Synthetic (conjugation) reactions. Couple a drug or Phase I metabolite with an endogenous substrate (glucuronide, sulphate, glycine, acetate, methyl) to form water-soluble conjugate for excretion.
Metabolites usually inactive (exception: morphine-6-glucuronide is more potent than morphine).
CARD 28
Q: Phase II reactions are usually _____ than Phase I. Metabolites are mostly _____.
A: Faster than Phase I. Metabolites mostly inactive.
CARD 29
Q: Can Phase II precede Phase I? Give example.
A: YES. Isoniazid (INH): first acetylated (Phase II) → then hydrolysed (Phase I) to isonicotinic acid.
CARD 30
Q: Phase I drugs - CYP450 dependent oxidation examples (circle-marked in your book)?
A: Phenobarbital, Phenytoin, Carbamazepine, Morphine, Codeine, Propranolol, Acetaminophen, Diazepam
CARD 31
Q: Phase I reduction examples?
A: Chloramphenicol, clonazepam, dantrolene, methadone, naloxone
CARD 32
Q: Phase I hydrolysis examples?
A: Procaine, lignocaine, procainamide, aspirin, indomethacin, succinylcholine
CARD 33
Q: Phase II - Acetylation examples?
A: Isoniazid, sulphonamides, dapsone, hydralazine, procainamide
CARD 34
Q: Phase II - Methylation examples?
A: Adrenaline, Dopamine, Histamine
CARD 35
Q: Phase II - Glucuronide conjugation examples?
A: Diazepam, Digoxin, Digitoxin, morphine, meprobamate
CARD 36
Q: Phase II - Sulfation examples?
A: Methyldopa, acetaminophen
CARD 37
Q: Phase II - Glutathione conjugation examples?
A: Acetaminophen, ethacrynic acid
TOPIC 5: DRUG-METABOLISING ENZYMES
CARD 38
Q: Where are microsomal enzymes located? What are the most important ones?
A: In the endoplasmic reticulum of the liver and other tissues. Most important: Mixed-Function Oxidases (MFOs) / Monooxygenases.
Two key enzymes: Flavoprotein (NADPH-CYP450 reductase) + Hemoprotein (Cytochrome P450).
CARD 39
Q: Which CYP isoform metabolises >50% of prescription drugs?
A: CYP3A4 (accounts for ~36% of drug biotransformation)
CARD 40
Q: Enzyme induction: What is the main mechanism? How long does it take?
A: Increased synthesis of enzyme protein (mainly cytochrome P450 + glucuronyl transferase). Takes 4-14 days to reach peak. Levels return to normal 1-3 weeks after stopping inducer.
CARD 41
Q: Which CYP is induced by tobacco smoke + cruciferous vegetables?
A: CYP1A2 (also induced by omeprazole)
CARD 42
Q: Which CYP is induced by ethanol and isoniazid?
A: CYP2E1
CARD 43
Q: Which CYPs are induced by barbiturates and rifampicin?
A: CYP2A6, 2B6, 2C8, 2C9, and 2C19
CARD 44
Q: Which CYP is induced by rifampicin, phenobarbitone, phenytoin, carbamazepine, atorvastatin?
A: CYP3A4
CARD 45
Q: What is autoinduction? Example?
A: When an inducing drug increases its own metabolism. Examples: Rifampicin and carbamazepine can induce their own metabolism.
CARD 46
Q: Clinical implications of enzyme induction (5 points)?
A:
- Increased inactivation of simultaneously given drugs (e.g., rifampicin → contraceptive failure)
- Autoinduction (rifampicin, carbamazepine)
- Increased metabolism of endogenous substances (steroids, bilirubin)
- Phenytoin useful in Cushing's syndrome (increases cortisol metabolism)
- Phenobarbitone: clears congenital haemolytic jaundice; porphyrin synthesis increased by barbiturates (precipitates acute intermittent porphyria)
- Prodrugs are activated by enzyme induction
CARD 47
Q: What are suicide inhibitors of CYP? Examples?
A: Drugs that bind irreversibly to CYP450 apoprotein or heme by covalent bonding → permanent inhibition. Examples: Grapefruit juice, clopidogrel, ritonavir, propylthiouracil, ticlopidine.
CARD 48
Q: What is slow vs fast acetylator? Clinical significance?
A:
- Fast acetylators: Drug inactivated quickly → may need higher dose; reduced efficacy
- Slow acetylators: Drug inactivated slowly → risk of adverse effects
- Isoniazid → peripheral neuropathy in slow acetylators
- Hydralazine + procainamide → risk of lupus in slow acetylators
🎯 MCQ PRACTICE (30 Questions)
Q1. A 70 kg adult has a hepatic blood flow of 90 L/hour. A drug has hepatic clearance of 45 L/hour. What is its extraction ratio?
- A) 25%
- B) 50% ✅
- C) 75%
- D) 90%
ER = CL(liver)/Q = 45/90 = 0.5 = 50%
Q2. Which of the following has 100% oral bioavailability?
- A) Morphine
- B) Lignocaine
- C) Intravenous glucose ✅
- D) Propranolol
IV route = always 100% bioavailability
Q3. Atenolol has low oral bioavailability because it is:
- A) Too lipophilic
- B) Too hydrophilic ✅
- C) Highly protein bound
- D) A prodrug
Too hydrophilic → cannot cross lipid cell membrane
Q4. The bioavailability of different formulations in correct descending order is:
- A) Tablet > Suspension > Solution > Capsule
- B) Solution > Suspension > Tablet > Coated tablet > Capsule ✅
- C) Capsule > Coated tablet > Tablet > Suspension > Solution
- D) Solution > Tablet > Suspension > Capsule > Coated tablet
Q5. Amorphous chloramphenicol palmitate has better bioavailability than crystalline form because:
- A) It is more lipophilic
- B) It has smaller particle size
- C) It dissolves faster ✅
- D) It has lower plasma protein binding
Q6. Which drug is NOT used orally due to very high first-pass metabolism?
- A) Warfarin
- B) Phenytoin
- C) Morphine ✅
- D) Digoxin
Q7. First-pass metabolism involves metabolism in: (Select all correct)
- A) Gut wall ✅
- B) Portal blood ✅
- C) Liver ✅
- D) Kidney
All three - gut wall, portal blood, AND liver
Q8. Warfarin has aVd of 0.15 L/kg. This means:
- A) It is highly lipid soluble
- B) It is widely distributed to tissues
- C) It is highly plasma protein bound ✅
- D) It undergoes first-pass metabolism
Q9. Dialysis is NOT useful in toxicity of which drug?
- A) Lithium
- B) Warfarin ✅
- C) Phenobarbitone
- D) Salicylates
Warfarin is 90% protein bound → dialysis cannot filter it
Q10. Acidic drugs bind to:
- A) α1-acid glycoprotein
- B) Albumin ✅
- C) β-globulin
- D) γ-globulin
Acidic → albumin. Basic → α1-acid glycoprotein
Q11. Which of the following is a Phase I reaction?
- A) Glucuronidation
- B) Acetylation
- C) Sulfation
- D) Hydrolysis ✅
Q12. Phase II reactions are called synthetic because:
- A) They introduce functional groups
- B) They conjugate drug with endogenous substrate ✅
- C) They require NADPH
- D) They occur in ER
Q13. Morphine-6-glucuronide is notable because:
- A) It is less potent than morphine
- B) It is more potent than morphine ✅
- C) It is a Phase I metabolite
- D) It is formed by sulfation
Q14. Isoniazid is first ____ (Phase II), then ____ (Phase I):
- A) Hydroxylated, then conjugated
- B) Acetylated, then hydrolysed ✅
- C) Hydrolysed, then acetylated
- D) Glucuronidated, then oxidised
Q15. Chloramphenicol is metabolised by which Phase I reaction?
- A) Oxidation
- B) Hydrolysis
- C) Reduction ✅
- D) Acetylation
Q16. Procaine is metabolised by which Phase I reaction?
- A) Oxidation
- B) Reduction
- C) Hydrolysis ✅
- D) Methylation
Q17. Which drugs undergo methylation (Phase II)?
- A) Isoniazid, sulphonamides
- B) Adrenaline, dopamine, histamine ✅
- C) Diazepam, digoxin
- D) Acetaminophen, ethacrynic acid
Q18. Microsomal enzymes are located in:
- A) Mitochondria
- B) Cytoplasm
- C) Endoplasmic reticulum ✅
- D) Golgi apparatus
Q19. CYP3A4 metabolises approximately what percentage of prescription drugs?
- A) 11%
- B) 19%
- C) 36% ✅ (over 50% of prescription drugs per your book text)
- D) 50%
Q20. Enzyme induction takes how long to reach peak effect?
- A) 1-2 days
- B) 4-14 days ✅
- C) 2-3 weeks
- D) 1-2 months
Q21. Rifampicin causes contraceptive failure due to:
- A) Inhibiting CYP3A4
- B) Inducing CYP3A4 → increases metabolism of oral contraceptives ✅
- C) Displacing contraceptive from protein binding
- D) Reducing GI absorption
Q22. Which drug can precipitate acute intermittent porphyria by inducing delta-ALA synthase?
- A) Rifampicin
- B) Barbiturates ✅
- C) Carbamazepine
- D) Phenytoin
Q23. CYP1A2 is induced by:
- A) Rifampicin
- B) Ethanol
- C) Tobacco smoke ✅
- D) Barbiturates
Q24. CYP2E1 is induced by:
- A) Tobacco smoke
- B) Ethanol + Isoniazid ✅
- C) Rifampicin
- D) Omeprazole
Q25. Which is an example of autoinduction?
- A) Rifampicin causing contraceptive failure
- B) Carbamazepine increasing its own metabolism ✅
- C) Phenytoin treating Cushing's syndrome
- D) Phenobarbitone clearing jaundice
Q26. Slow acetylators are at risk of (isoniazid):
- A) Hepatotoxicity (fast acetylators risk this more)
- B) Peripheral neuropathy ✅
- C) Optic neuritis
- D) Drug resistance
Q27. Which of the following is a suicide inhibitor of CYP?
- A) Rifampicin
- B) Phenobarbitone
- C) Grapefruit juice ✅
- D) Carbamazepine
Q28. Phenytoin may be useful in Cushing's syndrome because:
- A) It inhibits cortisol synthesis
- B) It induces CYP enzymes → increases cortisol metabolism ✅
- C) It blocks cortisol receptors
- D) It increases ACTH
Q29. A patient on severe hepatic disease takes propranolol. What happens?
- A) Decreased bioavailability due to poor absorption
- B) Increased bioavailability due to reduced first-pass metabolism ✅
- C) No change in bioavailability
- D) Increased first-pass metabolism
Q30. Which of the following undergoes glucuronide conjugation (Phase II)?
- A) Isoniazid
- B) Adrenaline
- C) Digoxin ✅
- D) Methyldopa
Quick Summary Table (Stick on your desk tonight)
| Topic | Key Point |
|---|---|
| Bioavailability | AUC; IV=100%; F = f×(1-ER) |
| First-pass | Gut wall + Portal blood + Liver |
| Very high FPM (not oral) | Testosterone, Morphine, Isoprenaline, Lignocaine, Hydrocortisone |
| Formulation order | Solution > Suspension > Tab > Coated tab > Capsule |
| Warfarin | 90% PPB, aVd = 0.15 L/kg, no dialysis |
| Acidic drugs | Bind albumin |
| Basic drugs | Bind α1-acid glycoprotein |
| Phase I | Non-synthetic; -OH/-SH/-NH2; Oxidation/Reduction/Hydrolysis |
| Phase II | Synthetic/Conjugation; mostly inactive products |
| CYP3A4 | Biggest isoform (36%); induced by rifampicin |
| CYP1A2 | Induced by tobacco smoke |
| CYP2E1 | Induced by ethanol + isoniazid |
| Enzyme induction | Peaks in 4-14 days |
| Slow acetylators | INH → neuropathy; Hydralazine → lupus |
| Isoniazid metabolism | Phase II (acetylation) FIRST, then Phase I (hydrolysis) |
Good luck tomorrow! Cover these 30 MCQs first, then scan the quick summary table right before you start micro and patho. 💪
This is a shared conversation. Sign in to Orris to start your own chat.