Young onset dementia for viva crisp and important things with flow chart manner
Reading File
Finding Sources
Finding Sources
Searching PubMed
"young onset dementia" diagnosis management
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
I now have comprehensive textbook data. Let me compile the full viva-ready flowchart-style answer.
Young-Onset Dementia (YOD) — Viva Crisp Notes
DEFINITION
| Term | Age cut-off |
|---|---|
| Young-onset dementia (YOD) | < 45 years |
| Early-onset dementia | < 65 years |
| Late-onset dementia | ≥ 65 years |
The boundary is not universal — some authorities use <65 for "young-onset." Know both and state what definition you are using.
EPIDEMIOLOGY
- Prevalence ≈ 67–81/100,000 persons aged 45–64
- Represents ~5–9% of all dementia cases
- Often delayed diagnosis (mean delay 4–5 years) due to atypical presentations and low clinical suspicion
CAUSES — FLOWCHART BY AGE
DEMENTIA ONSET
│
├─── Age < 45 yrs (Kelley 2008, Mayo Clinic)
│ Most common:
│ 1. Frontotemporal dementia (FTD)
│ 2. Huntington disease
│ 3. Multiple sclerosis
│ 4. Autoimmune/inflammatory encephalopathy
│ 5. Neuropsychiatric lupus
│ 6. Mitochondrial disease
│ 7. Lysosomal storage disease
│ 8. Prion disease (CJD)
│ 9. CNS vasculitis
│
└─── Age 30–65 yrs (Harvey 2003)
Most common:
1. Alzheimer dementia (most common >45)
2. Vascular dementia
3. Frontotemporal dementia
4. Alcohol-related dementia
5. Dementia with Lewy bodies (DLB)
6. Huntington disease
7. Multiple sclerosis
8. Down syndrome dementia
9. CBD / prion disease / Parkinson dementia
Mayo Clinic overall breakdown: Neurodegenerative 31.1% → Autoimmune/Inflammatory 21.3% → Metabolic 10.6% → Unknown 18.7%
BROAD AETIOLOGICAL FRAMEWORK
YOD CAUSES
├── NEURODEGENERATIVE
│ ├── Alzheimer disease (dominant if >45)
│ ├── Frontotemporal dementia (dominant if <45)
│ ├── Dementia with Lewy bodies
│ ├── Corticobasal degeneration (CBD)
│ └── Huntington disease (CAG repeat ↑)
│
├── VASCULAR
│ ├── Multi-infarct dementia
│ ├── Binswanger disease (subcortical)
│ └── CADASIL (Notch3 mutation — classic YOD cause)
│
├── AUTOIMMUNE / INFLAMMATORY
│ ├── Autoimmune encephalitis (anti-NMDAR, VGKC, LGI1)
│ ├── CNS vasculitis
│ ├── Neuropsychiatric SLE
│ └── MS / neurosarcoidosis
│
├── METABOLIC / TOXIC
│ ├── Wernicke-Korsakoff (thiamine ↓)
│ ├── Vitamin B12 deficiency
│ ├── Wilson disease (copper)
│ ├── Mitochondrial disorders (MELAS)
│ ├── Alcohol-related
│ └── Drug toxicity (lithium, methotrexate)
│
├── INFECTIOUS
│ ├── HIV dementia
│ ├── Neurosyphilis
│ ├── Viral encephalitis
│ ├── Cryptococcal meningitis
│ ├── Tuberculosis
│ └── Whipple disease (PAS+ macrophages)
│
├── PRION
│ └── CJD (sporadic/variant/familial/iatrogenic)
│ — Rapidly progressive, myoclonus, EEG periodic complexes
│
├── NEOPLASTIC
│ ├── CNS lymphoma
│ ├── Gliomatosis cerebri
│ └── Paraneoplastic / cancer-associated encephalopathy
│
├── STRUCTURAL
│ ├── Normal-pressure hydrocephalus (triad: gait, incontinence, dementia)
│ ├── Chronic subdural hematoma
│ └── Intracranial tumours (meningioma)
│
└── STORAGE / GENETIC
├── Niemann-Pick type C
├── Metachromatic leukodystrophy
├── CADASIL
└── Down syndrome (Trisomy 21)
CLINICAL APPROACH — FLOWCHART
Patient < 65 yrs with cognitive decline
│
▼
EXCLUDE PSEUDODEMENTIA
Depression (masked depression in elderly)
Schizophrenia (usually younger, delusions prominent, memory intact)
│
▼
HISTORY
├── Pattern of onset (acute/subacute → vascular, autoimmune, prion)
├── Family history (Huntington, CADASIL, FAD, prion)
├── Drug/alcohol history
├── Systemic features (rash → SLE, chorea → Huntington)
└── Rate of progression (weeks–months = rapidly progressive dementia)
│
▼
EXAMINATION
├── Frontal lobe signs → FTD
├── Chorea → Huntington disease
├── Parkinsonism → DLB, CBD, PSP
├── Myoclonus → prion, metabolic
├── Cerebellar signs → MSA, mitochondrial, prion
└── Kayser-Fleischer rings → Wilson disease
│
▼
INVESTIGATIONS (TIERED)
INVESTIGATIONS — FLOWCHART
TIER 1 — ALL YOD PATIENTS (Minimum workup)
├── FBC, ESR, CRP
├── Metabolic panel (renal, LFTs, glucose, calcium)
├── TFTs (hypothyroidism → reversible)
├── Vitamin B12, folate, thiamine
├── HIV serology
├── VDRL/TPPA (neurosyphilis)
├── Lipid profile
└── MRI brain (mandatory — rule out structural/vascular/atrophy pattern)
TIER 2 — BASED ON HISTORY/SUSPICION
├── EEG (non-convulsive status, prion)
├── Genetic testing: HTT CAG repeat (Huntington), NOTCH3 (CADASIL)
├── Autoimmune panel: ANA, ANCA, anti-dsDNA, complement
├── Paraneoplastic antibodies (anti-Hu, Yo, Ri, NMDAR, LGI1, CASPR2)
├── Copper/ceruloplasmin/24hr urine copper (Wilson disease)
├── Lactate/pyruvate, mitochondrial DNA (mitochondrial disease)
├── Lysosomal enzyme assays (storage diseases)
├── Chest X-ray / CT chest (sarcoidosis, TB, malignancy)
└── CSF analysis:
├── Cell count, protein, glucose, VDRL
├── 14-3-3 protein, tau, RT-QuIC (prion)
├── Amyloid-β, phospho-tau (Alzheimer)
├── Oligoclonal bands (MS)
└── Cytology (CNS lymphoma, leptomeningeal)
TIER 3 — SPECIALIST/RESEARCH
├── PET: FDG-PET (metabolic pattern), amyloid PET, tau PET
├── SPECT: DaTscan (dopaminergic) for DLB
├── Brain biopsy (CNS vasculitis, lymphoma, prion if uncertain)
└── Skin biopsy (CADASIL — electron microscopy GOM deposits)
KEY DIAGNOSTIC CLUES — VIVA TRAPS
| Feature | Diagnosis |
|---|---|
| CAG repeat expansion (HTT gene) | Huntington disease |
| Chorea + dementia + psychiatric sx | Huntington disease |
| NOTCH3 mutation + migraines + white matter lesions | CADASIL |
| Rapid progression + myoclonus + periodic EEG | CJD (prion) |
| RT-QuIC positive CSF | Prion disease (high specificity) |
| 14-3-3 protein in CSF | CJD (sensitive, not specific) |
| Kayser-Fleischer rings + liver disease | Wilson disease |
| KF rings + low ceruloplasmin | Wilson disease |
| Anti-NMDAR antibodies | Autoimmune encephalitis |
| Personality change + disinhibition + preserved memory early | FTD |
| Fluctuating cognition + visual hallucinations + parkinsonism | DLB |
| Gait apraxia + urinary incontinence + dementia | Normal-pressure hydrocephalus |
| PAS+ macrophages in gut biopsy | Whipple disease |
| Maternal inheritance + lactic acidosis + stroke-like episodes | MELAS (mitochondrial) |
TREATABLE / REVERSIBLE CAUSES — MUST NOT MISS
REVERSIBLE YOD
├── Hypothyroidism
├── Vitamin B12 / thiamine deficiency
├── Wilson disease (penicillamine / trientine)
├── Neurosyphilis (penicillin)
├── HIV (antiretrovirals)
├── Autoimmune encephalitis (steroids, IVIG, plasma exchange)
├── CNS vasculitis (immunosuppression)
├── Normal-pressure hydrocephalus (VP shunt)
├── Chronic subdural haematoma (drainage)
├── CNS lymphoma (chemotherapy ± radiotherapy)
├── Cryptococcal meningitis (antifungals)
└── Drug toxicity (withdrawal of offending agent)
A key viva point: ~20% of YOD cases have potentially treatable aetiologies — this justifies a thorough workup in every case.
GENETIC YOD — SUMMARY TABLE
| Disease | Gene | Inheritance | Key feature |
|---|---|---|---|
| Huntington | HTT (CAG ≥ 36) | AD | Chorea, psychiatric sx |
| CADASIL | NOTCH3 | AD | Migraine, lacunar infarcts, white matter lesions |
| FAD (familial AD) | APP, PSEN1, PSEN2 | AD | Early amyloid accumulation |
| Prion (gCJD) | PRNP | AD/Sporadic | Rapid progression |
| Wilson disease | ATP7B | AR | Liver + neuro |
| MELAS | Mitochondrial DNA | Maternal | Lactic acidosis, stroke-like |
| Storage diseases | Various | AR | Multi-system, organomegaly |
MANAGEMENT FLOWCHART
DIAGNOSIS CONFIRMED
│
├─── SPECIFIC TREATMENT (if treatable cause)
│
├─── SYMPTOMATIC PHARMACOLOGICAL
│ ├── Cognitive: Cholinesterase inhibitors (AD, DLB)
│ ├── Behavioural: SSRIs (FTD, depression)
│ ├── Psychosis: Quetiapine (avoid typical antipsychotics in DLB)
│ └── Chorea: Tetrabenazine (Huntington)
│
├─── NON-PHARMACOLOGICAL
│ ├── Cognitive stimulation therapy
│ ├── Occupational therapy / ADL support
│ ├── Speech & language therapy
│ └── Carer education & support
│
├─── SOCIAL / LEGAL
│ ├── Driving cessation (early counselling)
│ ├── Power of attorney / advance directives
│ ├── Work/financial planning (younger patients still employed)
│ └── Child/family counselling (genetic implications)
│
└─── SPECIALIST REFERRAL
├── Neurologist (diagnosis, imaging, genetic)
├── Neuropsychologist (formal testing)
├── Geriatric psychiatry (BPSD management)
└── Genetic counsellor (hereditary causes)
VIVA KEY POINTS — CRISP ANSWERS
- Definition: YOD = onset < 65 yrs; strict definition < 45 yrs (Mayo/Kelley)
- Most common cause <45: Frontotemporal dementia + Huntington disease
- Most common cause 45–65: Alzheimer disease
- Most important distinction from late-onset: Higher proportion of treatable/reversible causes; genetic aetiology more prominent; social consequences more severe (employment, childcare, finances)
- Why investigate thoroughly?: ~10–20% have treatable causes; missing them is catastrophic
- Fastest to diagnose (must not miss): Autoimmune encephalitis, neurosyphilis, B12/thiamine, hypothyroidism, NPH, subdural haematoma
- Rapidly progressive YOD → prion disease until proven otherwise; send RT-QuIC, 14-3-3, EEG
- CSF biomarkers in AD: ↑ p-tau, ↑ t-tau, ↓ Aβ42
- DLB: Never give typical antipsychotics — risk of severe neuroleptic sensitivity reaction
- FTD vs AD: FTD = personality/behaviour change first, memory relatively preserved early; AD = memory loss first
Sources: Bradley and Daroff's Neurology in Clinical Practice; Neuroanatomy through Clinical Cases, 3rd Ed; Adams and Victor's Principles of Neurology, 12th Ed; Kaplan & Sadock's Comprehensive Textbook of Psychiatry
Approach to inflammatory myopathies
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Excellent — I now have comprehensive data from multiple authoritative sources. Let me compile the full viva answer.
Approach to Inflammatory Myopathies — Viva Crisp Notes
CLASSIFICATION (Current Framework)
INFLAMMATORY MYOPATHIES (IMs)
├── Dermatomyositis (DM) — including Juvenile DM
├── Polymyositis (PM) — rare as isolated entity
├── Immune-Mediated Necrotizing Myopathy (IMNM)
├── Antisynthetase Syndrome (ASyS)
├── Inclusion Body Myositis (IBM)
└── Others:
├── Overlap myositis (with CTD)
├── Checkpoint inhibitor myositis
├── Infectious myositis
├── Eosinophilic / Granulomatous myositis
└── Paraneoplastic myositis
True isolated PM without overlap signs is now considered rare — many previously labelled PM were actually IMNM, IBM, or muscular dystrophies.
EPIDEMIOLOGY AT A GLANCE
| Feature | DM | PM | IMNM | IBM |
|---|---|---|---|---|
| Sex | F > M | F > M | M = F | M > F |
| Age | Childhood + adult | Adult | All ages | >50 yrs (most common myopathy >50) |
| Rash | Yes | No | No | No |
| CK | Normal–↑↑ | ↑↑↑ | ↑↑↑↑ (>10× ULN) | Normal or mildly ↑ |
| Response to IS | Yes | Yes | Poor monotherapy | No |
STEP-BY-STEP CLINICAL APPROACH
PATIENT WITH MUSCLE WEAKNESS
│
▼
STEP 1: LOCALISE THE LESION
├── UMN signs? → CNS
├── Wasting + fasciculations? → Anterior horn cell / MND
├── Sensory loss + areflexia? → Peripheral nerve
├── Ptosis + fatigability? → NMJ (MG)
└── Proximal weakness, NO sensory loss → MYOPATHY
│
▼
STEP 2: IS IT INFLAMMATORY?
├── Subacute onset
├── Elevated CK
├── Elevated ESR/CRP
├── Skin rash (heliotrope, Gottron's)
├── Systemic features (ILD, arthritis, Raynaud's)
└── Auto-antibodies positive
│
▼
STEP 3: WHICH SUBTYPE?
SUBTYPE DIFFERENTIATION — FLOWCHART
SUSPECTED INFLAMMATORY MYOPATHY
│
┌─────────┴─────────┐
│ │
Proximal > Distal Finger flexor + Quadriceps weakness
weakness (distal + proximal, asymmetric)
│ │
▼ ▼
Has RASH? Suspect IBM
│ → See IBM pathway
┌─┴─┐
Yes No
│ │
▼ ▼
DM PM / IMNM / ASyS
│
Has anti-HMGR or anti-SRP?
│ │
Yes No
│ │
IMNM Anti-tRNA synthetase?
(anti-Jo-1, PL-7, PL-12, EJ, OJ)
│ │
Yes No
│ │
ASyS PM (or overlap)
DERMATOMYOSITIS (DM)
Skin Features — Must Know
| Sign | Description |
|---|---|
| Heliotrope rash | Violaceous rash around eyelids ± periorbital oedema |
| Gottron's papules | Erythematous papules over knuckles (MCP, PIP, DIP) — pathognomonic |
| Gottron's sign | Erythema over extensor surfaces (elbows, knees) |
| Shawl sign | V-neck / shawl distribution rash |
| Mechanic's hands | Hyperkeratotic fissuring of lateral fingers (also in ASyS) |
| Periungual changes | Dilated nail-fold capillaries, ragged cuticles |
| Calcinosis | More in juvenile DM |
Pathology
- Perimysial + perivascular inflammation
- Inflammatory infiltrate: CD4+ T cells, B cells, dendritic cells, macrophages
- MAC (complement C5b-9) deposition on capillaries
- MxA expression on fibres — marker of type-I interferon signature (DM-specific)
- Perifascicular atrophy — hallmark
MSAs in DM
| Antibody | Associated Features |
|---|---|
| Anti-Mi-2 | Classic DM, mild, good prognosis — less likely to need 2nd-line agent |
| Anti-TIF1-γ (anti-p155/140) | Malignancy-associated DM (especially in adults) |
| Anti-NXP2 | DM + calcinosis, malignancy in adults |
| Anti-MDA5 | Rapidly progressive ILD, minimal muscle weakness, skin ulcers |
| Anti-SAE | DM, dysphagia prominent |
POLYMYOSITIS (PM)
- Proximal > distal symmetric weakness, no rash
- Endomysial inflammation (vs perimysial in DM)
- Infiltrate: CD8+ T cells + macrophages (cytotoxic T-cell mediated — invade non-necrotic fibres)
- MHC-I ubiquitously upregulated on muscle fibres
- Diagnose by exclusion — must rule out IBM, IMNM, muscular dystrophy
- ⚠️ Many "PM" diagnoses historically were actually IMNM or IBM
IMMUNE-MEDIATED NECROTIZING MYOPATHY (IMNM)
Key Features
- Acute/insidious proximal > distal weakness
- CK markedly elevated (>10× ULN, often 5000–50,000 U/L)
- Minimal inflammatory infiltrate on biopsy — predominantly necrotic fibres
- MAC deposition on non-necrotic fibres + MHC-I upregulation
Two MSA-defined subtypes
| Antibody | Trigger | Key feature |
|---|---|---|
| Anti-HMGR | Statin use (>50 yrs) — also occurs in young without statins | Does NOT improve on statin cessation; can mimic LGMD |
| Anti-SRP | Idiopathic | Subacute, aggressive, refractory course |
Statin toxic myopathy: improves with cessation. Anti-HMGR IMNM: does NOT improve — requires immunotherapy
- 90% require 2nd-line agent within 6 months — start early
- Anti-HMGR IMNM: IVIG may work as monotherapy (useful if steroids contraindicated)
ANTISYNTHETASE SYNDROME (ASyS)
The Antisynthetase Triad (Classic)
- Inflammatory myopathy
- Interstitial lung disease (ILD) — can be life-threatening
- Inflammatory arthritis
Additional Features
- Mechanic's hands (hyperkeratotic, fissured lateral fingers)
- Raynaud's phenomenon
- Fever
- Anti-Jo-1 most common (others: anti-PL-7, PL-12, EJ, OJ, KS)
Anti-MDA5 ≠ ASyS — anti-MDA5 is a DM-MSA with amyopathic DM + rapidly progressive ILD
- ASyS frequently fails corticosteroid monotherapy — early 2nd-line agent needed
INCLUSION BODY MYOSITIS (IBM)
Clinical Pearls
- Most common myopathy in >50 years; M > F
- Distal + proximal involvement — asymmetric
- Characteristic pattern: deep finger flexors (flexor pollicis longus) + quadriceps
- Wrist/finger flexors > extensors
- Facial weakness + dysphagia in 1/3
- No myalgia
- Mimics MND (asymmetry + large MUPs on EMG) — but no fasciculations, no intrinsic hand wasting, reflexes preserved/reduced
Biopsy Hallmarks
- Endomysial inflammation + CD8+ T-cell invasion (similar to PM)
- Rimmed vacuoles (red rim on modified Gomori trichrome)
- TDP-43 inclusions — very specific
- p62 inclusions — very sensitive
- EM: tubulofilamentous inclusions 15–21 nm diameter
Antibody
- Anti-cN-1A (anti-NT5c1A): present in ~70% — specificity >90% for IBM vs other myopathies
Treatment
- No disease-modifying treatment proven effective
- Prednisone, IVIG, MTX, alemtuzumab — all negative in trials
- Management = supportive: fall prevention, ankle supports, gait aids, tendon transfer for hand function
INVESTIGATIONS FLOWCHART
INVESTIGATIONS
│
├── BLOODS
│ ├── CK — key screening + monitoring marker
│ ├── LDH, AST, ALT (muscle enzyme panel)
│ ├── ESR, CRP, CBC
│ ├── ANA (screen for CTD overlap)
│ └── Myositis-specific antibodies (MSA) + myositis-associated antibodies (MAA)
│
├── EMG/NCS
│ ├── Fibrillation potentials + PSWs (irritable myopathy)
│ ├── Small short polyphasic MUPs (myopathic)
│ ├── IBM: MIXED pattern (small + large MUPs — mimics neurogenic)
│ └── Guides biopsy site (avoid EMG'd muscle)
│
├── MRI MUSCLE (STIR)
│ ├── Oedema/inflammation = bright signal
│ ├── PM: rectus femoris prominent
│ ├── IBM: vastus lateralis/medialis with relative sparing of rectus femoris
│ └── Guides biopsy to most active area
│
├── MUSCLE BIOPSY — Gold Standard
│ ├── DM: perimysial + perivascular; perifascicular atrophy; MAC on capillaries
│ ├── PM: endomysial CD8+ T cells; MHC-I expression
│ ├── IMNM: necrosis >> inflammation; MAC on non-necrotic fibres
│ ├── IBM: rimmed vacuoles; TDP-43; tubulofilaments on EM
│ └── SKIN BIOPSY: for DM diagnosis (± muscle biopsy)
│
├── PULMONARY
│ ├── CXR + HRCT chest (ILD screen — all new IMs)
│ └── PFTs (FVC, DLCO)
│
└── MALIGNANCY SCREEN (all adult DM, especially anti-TIF1-γ)
├── CT chest/abdomen/pelvis
├── FDG-PET (increasing utility)
├── PSA, mammogram, colonoscopy (age/sex appropriate)
└── Repeat at 3 years (malignancy can precede or follow DM)
TREATMENT FLOWCHART
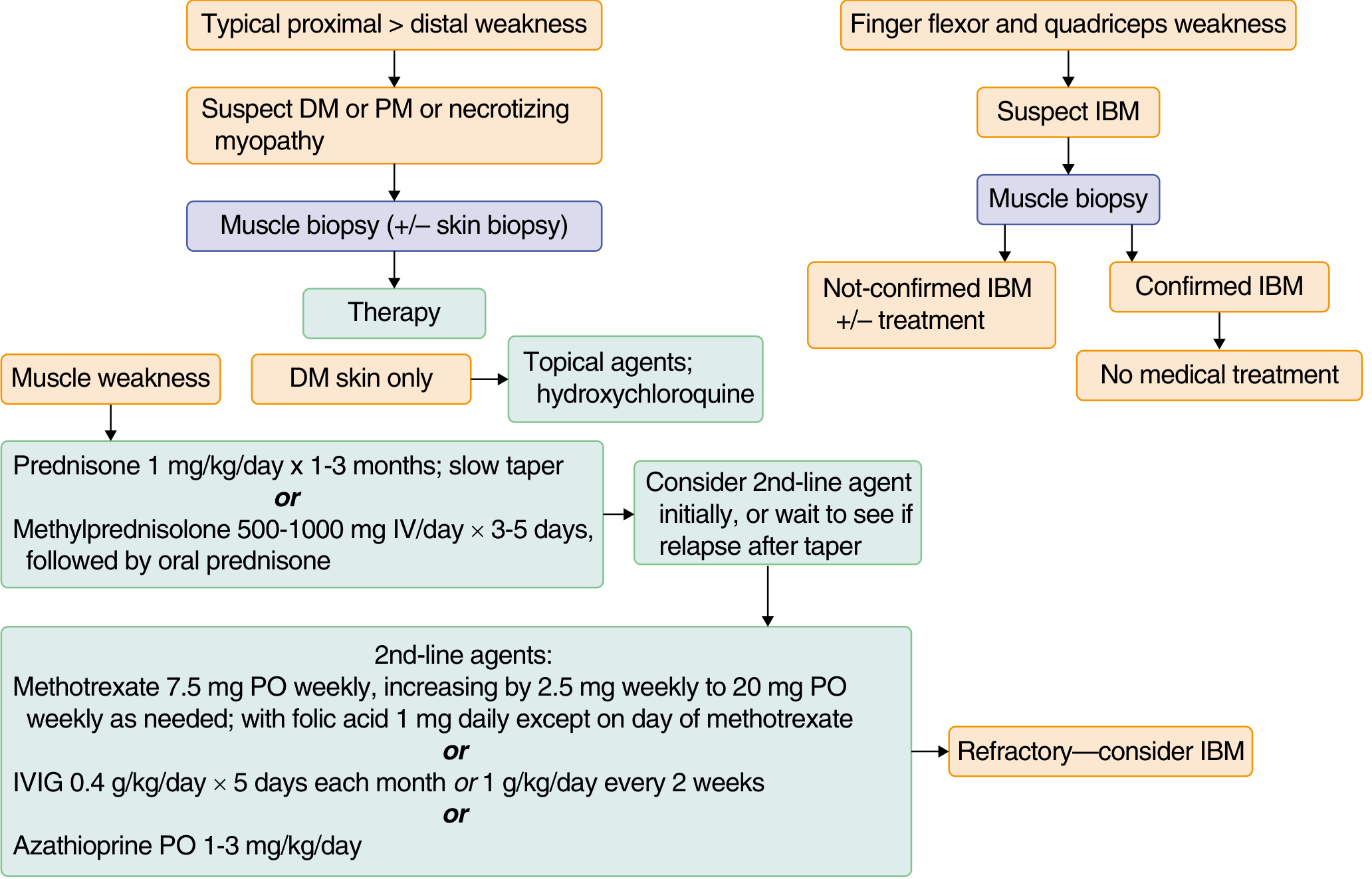
Treatment Summary
DM / PM / IMNM WITH MUSCLE WEAKNESS
│
├── FIRST LINE
│ Prednisone 0.7–1 mg/kg/day (up to 60 mg/day) × 2–4 months
│ OR IV methylprednisolone 1 g/day × 3–5 days (severe disease)
│ → Maintain until strength improves + CK normalises
│ → Then slow taper: 10 mg/month → reach 20 mg → taper more slowly
│ → Monitor: glucose, K+, BP, eyes annually, bones (Ca + Vit D)
│
├── SECOND LINE (add if: poor response 2–4 months, relapse on taper, steroid toxicity)
│ ├── Methotrexate: 7.5 mg/week → titrate to 20 mg/week + folic acid 1 mg/day
│ ├── Azathioprine: 1–3 mg/kg/day
│ ├── Mycophenolate mofetil: 2–3 g/day
│ └── IVIG: 2 g/kg over 2–5 days (Grade A evidence in DM)
│
├── THIRD LINE / REFRACTORY
│ ├── Rituximab (anti-CD20)
│ ├── Cyclophosphamide (severe ILD)
│ ├── Tacrolimus / ciclosporin
│ └── Plasma exchange
│
├── DM SKIN ONLY
│ ├── Topical steroids
│ ├── Hydroxychloroquine / chloroquine
│ └── Sun protection
│
└── IBM
└── SUPPORTIVE ONLY (no immunotherapy shown effective)
├── Avoid falls
├── AFOs / gait aids
└── Tendon transfer (hand function)
Antibody-guided treatment
| Antibody | Treatment implication |
|---|---|
| Anti-Mi-2 | Less likely to need 2nd-line agent |
| Anti-Jo-1 (ASyS) | Frequently fails steroid monotherapy — add 2nd-line early |
| Anti-HMGR (IMNM) | Start 2nd-line immediately OR within 1 month; IVIG may work as monotherapy |
| Anti-SRP (IMNM) | Aggressive, refractory — early combination therapy |
| Anti-MDA5 | Rapidly progressive ILD — early cyclophosphamide + steroids |
| Anti-TIF1-γ | Malignancy screen mandatory |
MALIGNANCY ASSOCIATION — VIVA MUST
- DM: 18–32% associated with malignancy (highest risk 3 yrs before/after diagnosis)
- PM: ~10–15%
- IBM: No significant malignancy association
- IMNM: can be paraneoplastic (anti-SRP less so; HMGR less so)
- Most common cancers: ovary, lung, GI, breast, lymphoma
- Anti-TIF1-γ = strongest malignancy predictor in adult DM
SYSTEMIC COMPLICATIONS
| System | Complication |
|---|---|
| Lung | ILD (ASyS, DM anti-MDA5, IMNM) — major cause of death |
| Heart | Myocarditis, arrhythmia, heart block |
| Joints | Inflammatory arthritis (ASyS) |
| Oesophagus | Dysphagia (proximal — striated muscle), aspiration pneumonia |
| Skin | Calcinosis (juvenile DM), ulcers (anti-MDA5) |
| Kidney | Rarely direct; drugs (MTX, CsA) |
KEY VIVA TRAPS
- IBM vs PM on biopsy: Both show endomysial CD8+ infiltrate — rimmed vacuoles + TDP-43 = IBM
- IBM vs MND: IBM has finger flexor > extensor weakness, no true fasciculations, no UMN signs, no intrinsic hand wasting
- IMNM is NOT the same as toxic statin myopathy — toxic myopathy resolves on stopping statin; IMNM persists and needs immunotherapy
- Anti-MDA5 = amyopathic DM + rapidly progressive ILD — muscle CK may be normal
- Perifascicular atrophy = DM even without inflammation visible on biopsy
- MAC on capillaries = DM; MAC on non-necrotic fibres = IMNM
- DM infiltrate = CD4+ / B cells / macrophages; PM/IBM = CD8+ T cells (cytotoxic)
- IVIG has Grade A RCT evidence in DM (NEJM 2022 trial — Aggarwal et al.)
- IBM never responds to immunotherapy — do not treat with steroids expecting benefit
- Juvenile DM: Calcinosis is prominent; vasculitis more common; skin can precede weakness by months
Sources: Harrison's Principles of Internal Medicine 22E; Bradley and Daroff's Neurology in Clinical Practice; Goldman-Cecil Medicine
This is a shared conversation. Sign in to Orris to start your own chat.