Transamination and delamination
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I have comprehensive content from authoritative biochemistry textbooks. Here is the full, detailed answer:
Transamination and Deamination
These two reactions are the primary ways the body removes nitrogen from amino acids, and they work sequentially - transamination first funnels nitrogen into glutamate, then deamination releases it as free ammonia.
1. Transamination
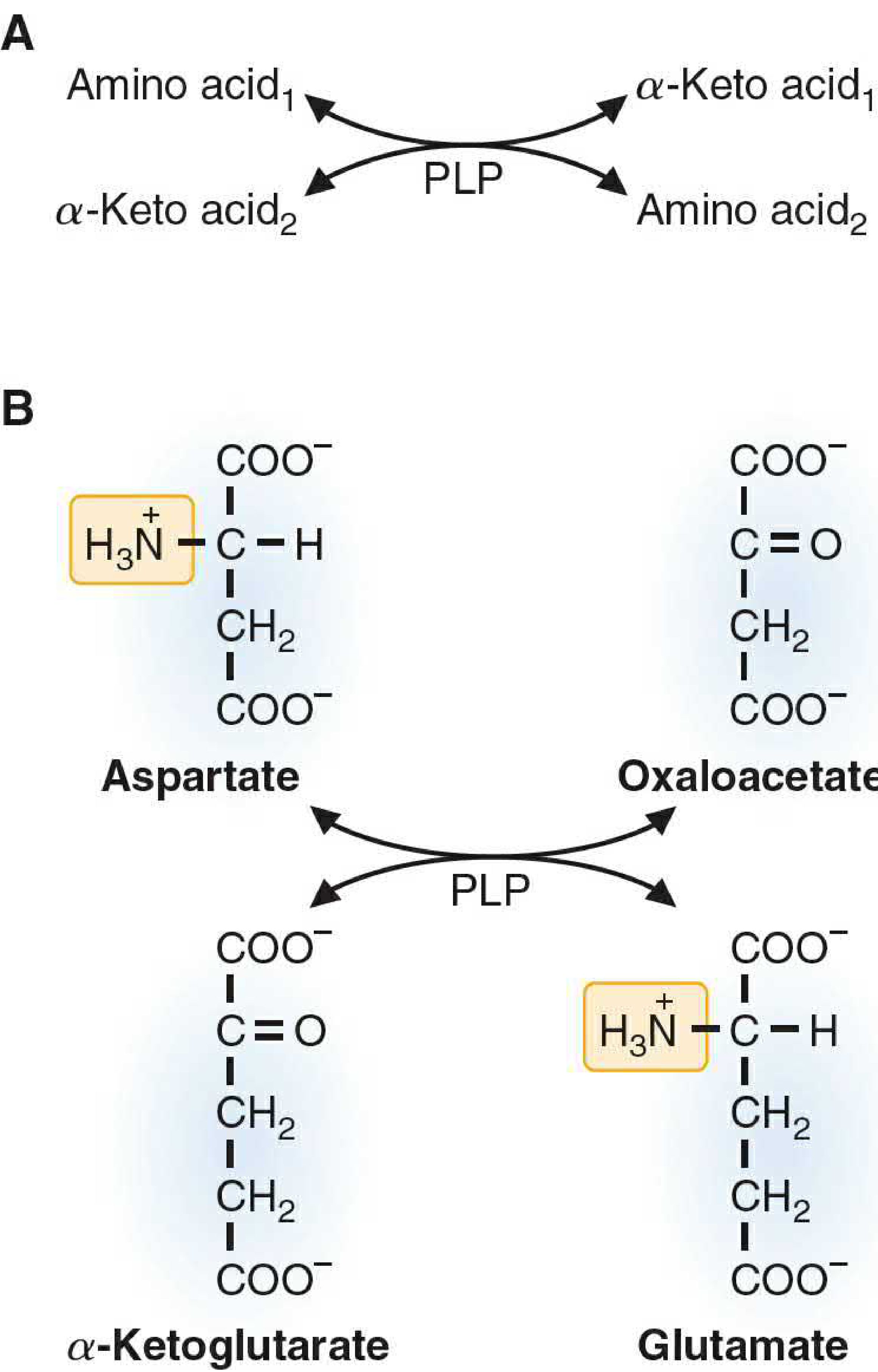
Definition: The transfer of an amino (-NH₂) group from an amino acid to an α-keto acid, converting the donor into its corresponding α-keto acid while the acceptor becomes an amino acid.
Key Features
| Feature | Detail |
|---|---|
| Enzyme | Aminotransferases (transaminases) |
| Cofactor | Pyridoxal phosphate (PLP) - derived from Vitamin B6 |
| Common acceptor | α-Ketoglutarate → glutamate |
| Reversibility | Freely reversible (equilibrium constant ~1) |
| Mechanism | "Ping-pong" (alternating substrate addition and product release) |
| Exceptions | Lysine and threonine do NOT undergo transamination (Harper's also adds proline and hydroxyproline) |
Prototypical Reactions
-
Aspartate aminotransferase (AST / GOT): Aspartate + α-Ketoglutarate ⇌ Oxaloacetate + Glutamate
-
Alanine aminotransferase (ALT / GPT): Alanine + α-Ketoglutarate ⇌ Pyruvate + Glutamate
Why α-Ketoglutarate/Glutamate is Central
Because virtually all aminotransferases use α-ketoglutarate as the nitrogen acceptor, the amino groups from most amino acids ultimately concentrate in glutamate. This is physiologically critical: glutamate is the only amino acid that undergoes oxidative deamination at an appreciable rate, linking transamination to ammonia production.
Mechanism (PLP as Nitrogen Carrier)
PLP sits at the active site covalently bound to the enzyme's lysine residue (as a Schiff base). During transamination:
- The amino acid displaces lysine, forming a Schiff base with PLP (external aldimine).
- The Schiff base rearranges (tautomerization) to a ketimine.
- Hydrolysis releases the α-keto acid and leaves PLP in the pyridoxamine phosphate (PMP) form (now carrying the amino group).
- An α-keto acid (usually α-ketoglutarate) reacts with PMP, reverses the steps, picks up the NH₂ group, and is released as glutamate - regenerating enzyme-bound PLP.
Clinical Relevance
- AST and ALT are released into the blood when liver or cardiac cells are injured. Elevated serum levels are key markers of hepatocellular damage (hepatitis, cirrhosis) and myocardial infarction.
2. Deamination (Oxidative Deamination)
Definition: The removal of an amino group from glutamate as free ammonia (NH₃), with simultaneous oxidation of the carbon skeleton back to α-ketoglutarate. This is the main step that liberates nitrogen as free ammonia for urea synthesis.
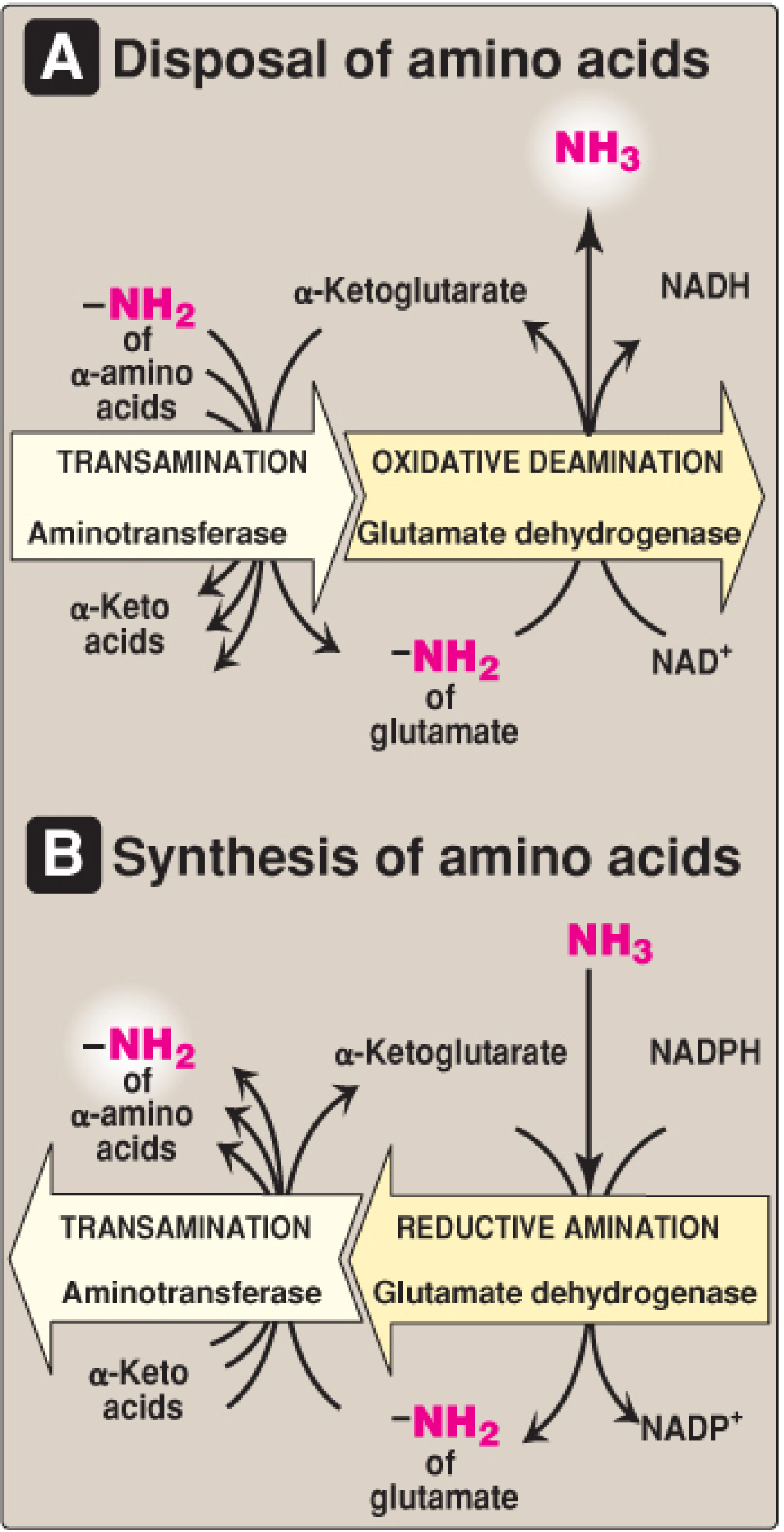
Glutamate Dehydrogenase (GDH) - The Key Enzyme
| Feature | Detail |
|---|---|
| Substrate | L-Glutamate |
| Location | Mitochondrial matrix (liver and kidney primarily) |
| Coenzyme | NAD⁺ (oxidative deamination) or NADPH (reductive amination) - unique dual specificity |
| Products | α-Ketoglutarate + NH₃ + NADH (or NADPH) |
| Activator | ADP (low energy signal) |
| Inhibitor | GTP (high energy signal) |
Reaction:
Glutamate + NAD⁺ → α-Ketoglutarate + NH₃ + NADH
Direction of Reaction
| Condition | Direction |
|---|---|
| After protein meal (high glutamate) | Forward → amino acid degradation → NH₃ production |
| High ammonia levels | Reverse → reductive amination → glutamate synthesis |
| Low energy (high ADP) | Forward (ADP activates GDH) |
| High energy (high GTP) | Reverse inhibited (GTP inhibits GDH) |
D-Amino Acid Oxidase (DAO)
A secondary deamination pathway in peroxisomes of liver and kidney:
- Acts on D-amino acids (from diet, not used in mammalian proteins)
- Flavin adenine dinucleotide (FAD)-dependent
- Produces α-keto acid + NH₃ + H₂O₂
- Increased DAO activity has been linked to susceptibility to schizophrenia (degrades D-serine, a modulator of NMDA receptors)
3. The Link Between Transamination and Deamination (Transdeamination)
These two processes act in sequence - a pathway sometimes called transdeamination:
Most amino acids
↓ (Aminotransferase + PLP)
Glutamate + α-keto acid
↓ (GDH + NAD⁺)
α-Ketoglutarate + NH₃ → enters urea cycle
This two-step sequence allows the amino nitrogen from virtually all amino acids (not just glutamate) to be funneled into the urea cycle. α-Ketoglutarate is regenerated in the second step and is recycled back into the first, making the whole system catalytic.
4. Ammonia Transport to the Liver
Since ammonia is toxic (especially to the brain), peripheral tissues do not release it directly. Two mechanisms carry nitrogen safely to the liver:
-
Glutamine shuttle: Glutamine synthetase combines NH₃ + glutamate → glutamine (non-toxic). In the liver, glutaminase cleaves it back to glutamate + NH₃, which then enters GDH.
-
Glucose-alanine cycle: Muscle transamination produces alanine from pyruvate. Alanine travels to the liver, ALT converts it back to pyruvate + glutamate; GDH then deaminates glutamate to release NH₃ for the urea cycle. The pyruvate is used for gluconeogenesis.
5. Summary Comparison
| Feature | Transamination | Oxidative Deamination |
|---|---|---|
| Nitrogen fate | Transferred to another α-keto acid | Released as free NH₃ |
| Enzyme | Aminotransferases | Glutamate dehydrogenase (GDH) |
| Cofactor | PLP (Vitamin B6) | NAD⁺ / NADPH |
| Location | Cytoplasm and mitochondria | Mitochondrial matrix |
| Reversibility | Freely reversible | Reversible (direction regulated by energy status) |
| Net result | No nitrogen loss; interconverts amino acids | Nitrogen leaves amino acid pool as NH₃ |
| Role | Funnels nitrogen into glutamate | Releases that nitrogen for urea synthesis |
Sources:
- Basic Medical Biochemistry: A Clinical Approach, 6e, pp. 1313-1315
- Lippincott's Illustrated Reviews: Biochemistry, 8e, pp. 706-710
- Harper's Illustrated Biochemistry, 32e, pp. 293-295
How does the urea cycle capture ammonia from GDH?
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have everything needed for a thorough answer. Here it is:
How the Urea Cycle Captures Ammonia from GDH
The handoff from GDH to the urea cycle is elegant - both processes share the same compartment (the mitochondrial matrix), so ammonia never has to travel far or enter the blood.
The Two Nitrogen Sources of Urea
Each urea molecule carries two nitrogen atoms. They come from different sources:
| Nitrogen atom | Source | Route |
|---|---|---|
| N-1 | Free NH₃ from GDH | → Carbamoyl phosphate → citrulline |
| N-2 | Amino group of aspartate | → Argininosuccinate → arginine |
Glutamate is the immediate precursor of both - it donates NH₃ via GDH (N-1) and donates its amino group to oxaloacetate via AST to make aspartate (N-2).
Step-by-Step: GDH → Urea Cycle
Here is the full cycle diagram showing exactly where and how ammonia enters:
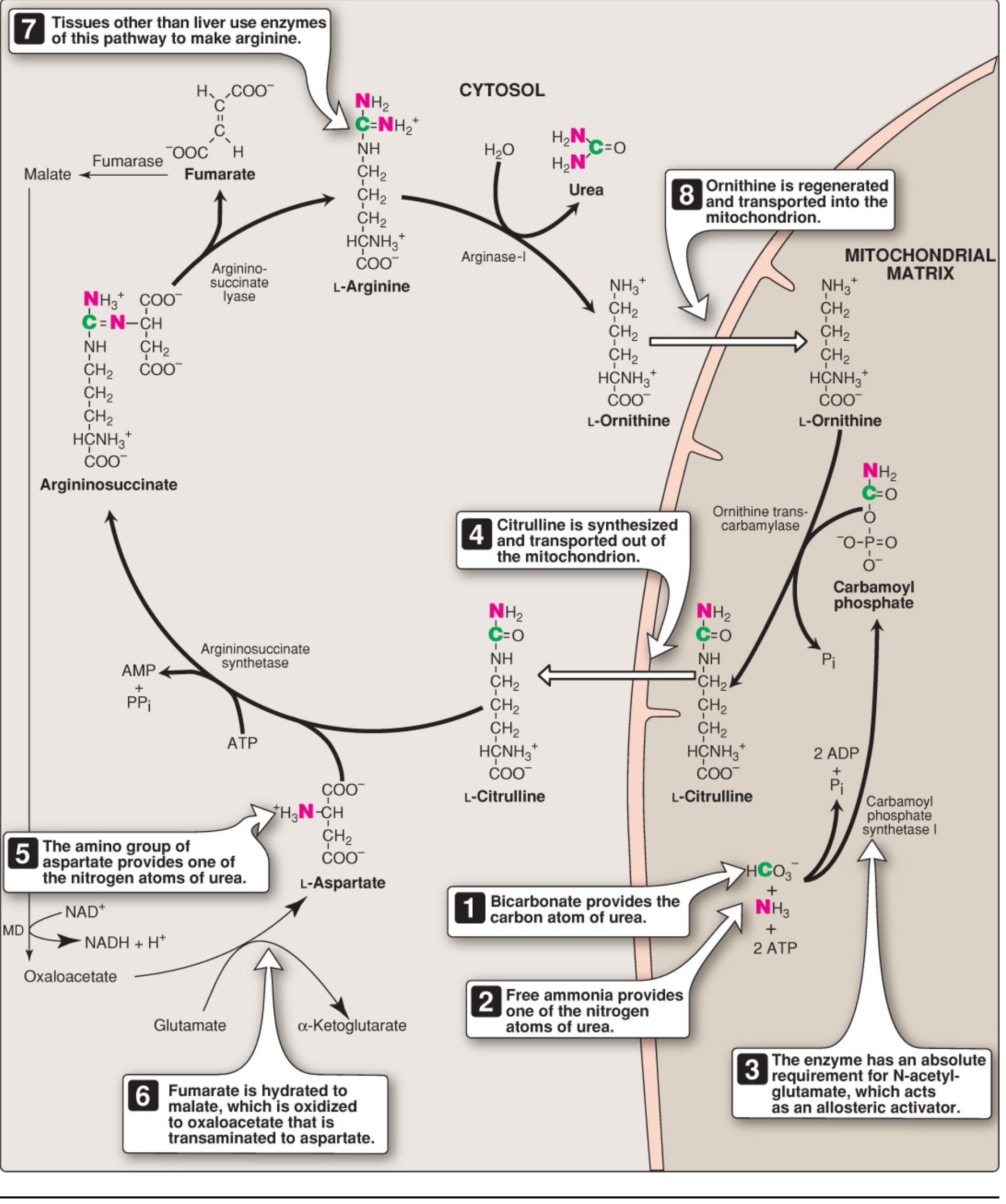
Step 1 - Carbamoyl Phosphate Synthesis (Mitochondrial Matrix)
GDH releases NH₃ → CPS-I immediately captures it.
NH₃ + HCO₃⁻ + 2 ATP ──CPS-I──→ Carbamoyl phosphate + 2 ADP + Pi
- Enzyme: Carbamoyl Phosphate Synthetase I (CPS-I) - located in the mitochondrial matrix, co-localised with GDH
- Energy cost: 2 ATP hydrolysed (one per phosphorylation step)
- Absolute requirement for allosteric activator: N-acetylglutamate (NAG)
- The ammonia incorporated here becomes one of the two nitrogens in urea
Why CPS-I and not CPS-II? CPS-II is the cytosolic isoform used for pyrimidine synthesis. It uses glutamine as the nitrogen source (not free NH₃) and does not require NAG. CPS-I is exclusively for the urea cycle.
Step 2 - Citrulline Formation (Still in Mitochondria)
Carbamoyl phosphate + Ornithine ──OTC──→ Citrulline + Pi
- Enzyme: Ornithine transcarbamylase (OTC) - mitochondrial
- The carbamoyl group (carrying GDH-derived nitrogen) is transferred to ornithine
- Citrulline is then exported to the cytosol via an antiporter that simultaneously imports ornithine
Steps 3-5 - Cytosolic Reactions (Adding the Second Nitrogen)
Citrulline + Aspartate ──Argininosuccinate synthetase──→ Argininosuccinate (costs ATP → AMP + PPi)
Argininosuccinate ──Argininosuccinate lyase──→ Arginine + Fumarate
Arginine ──Arginase-I──→ Urea + Ornithine
- Aspartate donates the second nitrogen (from its amino group) to form argininosuccinate, then arginine
- Arginase-I cleaves arginine, releasing urea (H₂N-CO-NH₂) and regenerating ornithine
- Ornithine is shuttled back into the mitochondria to start another round
The NAG "Throttle" on Ammonia Entry
The key regulatory point is CPS-I, controlled by N-acetylglutamate (NAG):
Glutamate + Acetyl-CoA ──NAG synthase──→ NAG
↑
Stimulated by Arginine
- NAG is synthesized specifically to activate CPS-I - it has no other known function in mammals
- Arginine (which builds up when amino acid catabolism is high) stimulates NAG synthesis, activating CPS-I AND producing more ornithine via arginase - a feed-forward amplification loop
- The overall cycle is regulated by substrate availability: the more NH₃ produced by GDH, the faster CPS-I runs
The "Krebs Bi-Cycle" Connection
The urea cycle and TCA cycle are physically linked via shared intermediates in the cytosol - sometimes called the Krebs bi-cycle:
- Fumarate released from argininosuccinate lyase → converted by fumarase to malate → oxidised to oxaloacetate
- Oxaloacetate is transaminated by AST to form aspartate (using glutamate as the nitrogen donor)
- This aspartate re-enters the urea cycle as the second nitrogen donor
- The NADH generated by malate dehydrogenase can feed the electron transport chain
This means the urea cycle is not just a detox loop - it is physically coupled to the TCA cycle, and the liver can run both simultaneously to process amino acids.
Why Compartmentalisation Matters
The physical co-location of GDH and CPS-I in the mitochondrial matrix is not accidental:
- NH₃ is membrane-permeable and toxic - if it were released into the cytosol or blood, it would cause damage
- By capturing it instantly with CPS-I in the same compartment, the liver keeps free ammonia concentrations extremely low (normal blood NH₃: 30-60 μM)
- If CPS-I fails (e.g. CPS-I deficiency or OTC deficiency), carbamoyl phosphate overflows into the cytosol and drives excess pyrimidine synthesis, producing orotic aciduria - a diagnostic clue
Clinical Consequence: When This Fails
When liver cells are destroyed (hepatic failure), GDH continues producing NH₃ from amino acids but CPS-I activity collapses. NH₃ enters the circulation and crosses the blood-brain barrier. In the brain:
- NH₃ + α-ketoglutarate → glutamate (via GDH) - depleting TCA intermediates
- Glutamate + NH₃ → glutamine - further draining α-ketoglutarate
- Result: impaired TCA cycle, reduced ATP, and hepatic encephalopathy (asterixis, disorientation, coma)
Sources:
- Basic Medical Biochemistry: A Clinical Approach, 6e, pp. 1321-1326
- Lippincott's Illustrated Reviews: Biochemistry, 8e, pp. 712-715
This is a shared conversation. Sign in to Orris to start your own chat.