Thrombosis
Reading File
Finding Sources
Searching PubMed
thrombosis pathophysiology treatment
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Here is a thorough overview of Thrombosis, drawn from Robbins & Kumar Basic Pathology and corroborated across multiple authoritative textbooks.
Thrombosis
Thrombosis is the formation of a solid mass of blood constituents - a thrombus - within an intact blood vessel or the heart during life. It represents one of the most common and serious pathological processes in clinical medicine, underlying myocardial infarction, stroke, pulmonary embolism, and deep vein thrombosis (DVT).
Virchow's Triad
The three primary abnormalities that lead to intravascular thrombosis are collectively called Virchow's Triad:
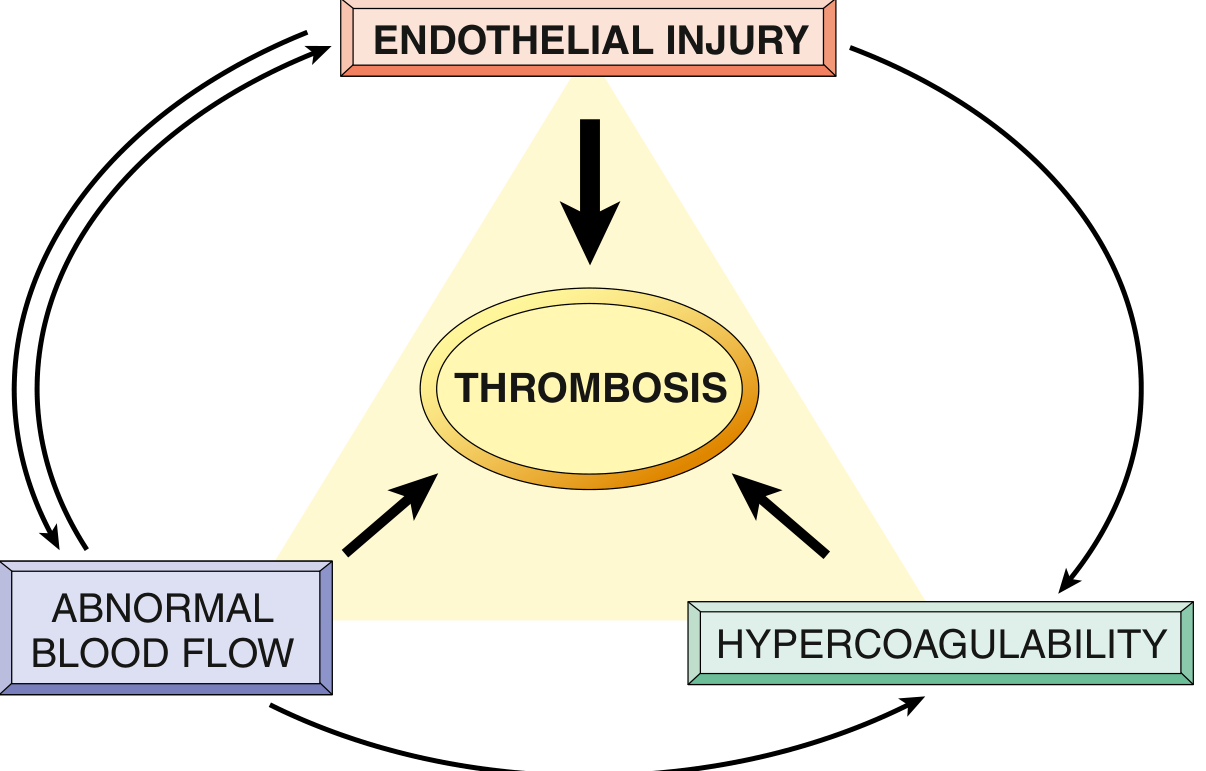
FIG. 3.12 - Virchow's Triad: Endothelial integrity is the most important factor. Abnormal blood flow (stasis or turbulence) can lead to hypercoagulability directly and also indirectly through endothelial dysfunction.
1. Endothelial Injury
Endothelial injury almost inevitably underlies thrombus formation in the heart and arterial circulation, where high flow rates otherwise prevent clotting. Arterial/cardiac thrombi are typically platelet-rich ("white thrombi").
Causes of endothelial injury/activation:
- Physical trauma, atherosclerotic plaque ulceration
- Infectious agents and cytokines
- Metabolic abnormalities: hypercholesterolemia, hyperhomocysteinemia
- Cigarette smoke toxins, hypertension
- Abnormal blood flow itself
Prothrombotic changes in activated endothelium:
- Procoagulant shift: downregulation of thrombomodulin, endothelial protein C receptor, and tissue factor pathway inhibitor (TFPI); upregulation of tissue factor
- Antifibrinolytic effects: increased secretion of PAI (plasminogen activator inhibitor), reducing t-PA activity
2. Abnormal Blood Flow (Stasis or Turbulence)
- Turbulence promotes arterial and cardiac thrombosis by causing endothelial dysfunction and creating local pockets of stasis
- Stasis is the dominant factor in venous thrombosis
Why stasis promotes thrombosis:
- Platelets and leukocytes contact the endothelium
- Activated clotting factors accumulate (impaired washout)
- Clotting factor inhibitors are not replenished
Clinical examples:
| Condition | Mechanism |
|---|---|
| Atherosclerotic plaques | Turbulence + endothelial injury |
| Aortic/arterial aneurysms | Local stasis |
| Atrial fibrillation + mitral stenosis | Atrial stasis |
| Post-MI ventricular aneurysm | Mural stasis |
| Polycythemia vera | Hyperviscosity - small vessel stasis |
3. Hypercoagulability
An abnormally high tendency of blood to clot, usually from altered coagulation factors. Divided into primary (genetic) and secondary (acquired) forms.
Primary (Genetic) Hypercoagulable States
| Disorder | Mechanism | Risk |
|---|---|---|
| Factor V Leiden mutation | Amino acid substitution renders factor V resistant to protein C cleavage | Heterozygotes: 3-4x increased VTE risk; Homozygotes: 25-50x increased risk |
| Prothrombin G20210A variant | 3'-UTR mutation → increased prothrombin expression | ~3x increased VTE risk |
| Antithrombin III deficiency | Loss of key coagulation inhibitor | Significant VTE risk |
| Protein C or S deficiency | Loss of anticoagulant pathway | VTE and warfarin-induced skin necrosis |
Secondary (Acquired) Hypercoagulable States
| High Risk | Lower Risk |
|---|---|
| Prolonged immobilization | Oral contraceptive use |
| Major surgery or trauma | Sickle cell disease |
| Malignancy (Trousseau syndrome) | Nephrotic syndrome |
| Antiphospholipid antibody syndrome | Hyperestrogenic states |
| HIT (heparin-induced thrombocytopenia) | Smoking |
Key acquired conditions:
-
HIT syndrome: Autoantibodies bind heparin-platelet factor 4 (PF4) complexes → platelet activation → paradoxical thrombosis despite low platelet counts. Occurs in up to 5% of patients on unfractionated heparin.
-
Antiphospholipid antibody syndrome (APS): Antibodies bind β2-glycoprotein I and phospholipid-associated proteins → recurrent arterial and venous thromboses, recurrent miscarriages, cardiac valve vegetations, thrombocytopenia. Paradoxically, these antibodies appear as anticoagulants in vitro but cause thrombosis in vivo.
Morphology of Thrombi
- Arterial thrombi: Attach to vessel wall, grow retrograde from attachment point; tend to be platelet-rich and pale ("white thrombi")
- Venous thrombi: Grow in the direction of blood flow (toward the heart); tend to be fibrin-rich and red ("red thrombi")
- Lines of Zahn: Pale layers of platelets + fibrin alternating with dark layers of red cells, seen in arterial thrombi and cardiac chambers - confirms ante-mortem formation
- Mural thrombi: Thrombi attached to the heart wall or aorta but not occlusive (e.g., overlying ventricular scars, in aortic aneurysms)
- Vegetations: Thrombi on cardiac valves - infected (infective endocarditis), sterile (non-bacterial thrombotic endocarditis in cancer/debilitating illness), or immune-mediated (Libman-Sacks in SLE)
Fates of Thrombi
Once formed, a thrombus has four possible fates:
- Propagation - grows larger by accumulating more platelets and fibrin
- Embolization - dislodges and travels to distant sites (most dangerous outcome)
- Dissolution (fibrinolysis) - t-PA activates plasmin which degrades fibrin. This is most effective when the thrombus is fresh. Therapeutic thrombolytics (e.g., t-PA) are generally not effective if given more than a few hours after thrombus formation.
- Organization and recanalization - older thrombi become organized by ingrowth of endothelial cells, smooth muscle cells, and fibroblasts. Capillary channels form, partially restoring lumen patency. The thrombus may ultimately be incorporated into the vessel wall as vascularized connective tissue.
Clinical Significance
| Location | Primary Risk |
|---|---|
| Deep veins (DVT) | Pulmonary embolism (most dangerous complication) |
| Coronary arteries | Myocardial infarction |
| Cerebral arteries | Ischemic stroke |
| Left heart / aorta | Arterial embolism → limb/visceral ischemia |
| Pulmonary arteries | Pulmonary hypertension (chronic PE) |
Most venous thrombi arise in the deep veins of the legs (DVT). DVTs that extend above the knee have a substantially higher risk of embolization than those confined to calf veins.
Summary Box (Robbins & Kumar)
Typically caused by one or more components of Virchow's triad: endothelial injury (e.g., by toxins, hypertension, inflammation, or metabolic products); static or turbulent blood flow (e.g., from aneurysms, atherosclerotic plaque); and hypercoagulability, either primary (genetic) or secondary (acquired). - Robbins & Kumar Basic Pathology
Sources: Robbins & Kumar Basic Pathology (9780323790185), Robbins, Cotran & Kumar Pathologic Basis of Disease (9780443264528), Sabiston Textbook of Surgery (9780443124341), Miller's Anesthesia (9780323935920)
Ischemia
Finding Sources
Searching PubMed
ischemia reperfusion injury mechanisms
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Here is a comprehensive overview of Ischemia, drawn from Robbins & Kumar Basic Pathology and Robbins, Cotran & Kumar Pathologic Basis of Disease.
Ischemia
Ischemia is a reduction or complete cessation of blood flow to a tissue, resulting in deprivation of both oxygen and metabolic substrates (glucose) while also preventing removal of waste products. This double insult makes ischemia more damaging than simple hypoxia alone.
Causes of Ischemia
| Category | Examples |
|---|---|
| Arterial occlusion | Thrombosis, embolism, atherosclerotic plaque |
| Venous occlusion | Venous thrombosis (prevents drainage, impairs inflow) |
| Reduced cardiac output | Cardiogenic shock, heart failure |
| Vasospasm | Coronary vasospasm (Prinzmetal angina) |
| Extrinsic compression | Tumor, torsion, compartment syndrome |
Mechanisms of Ischemic Cell Injury
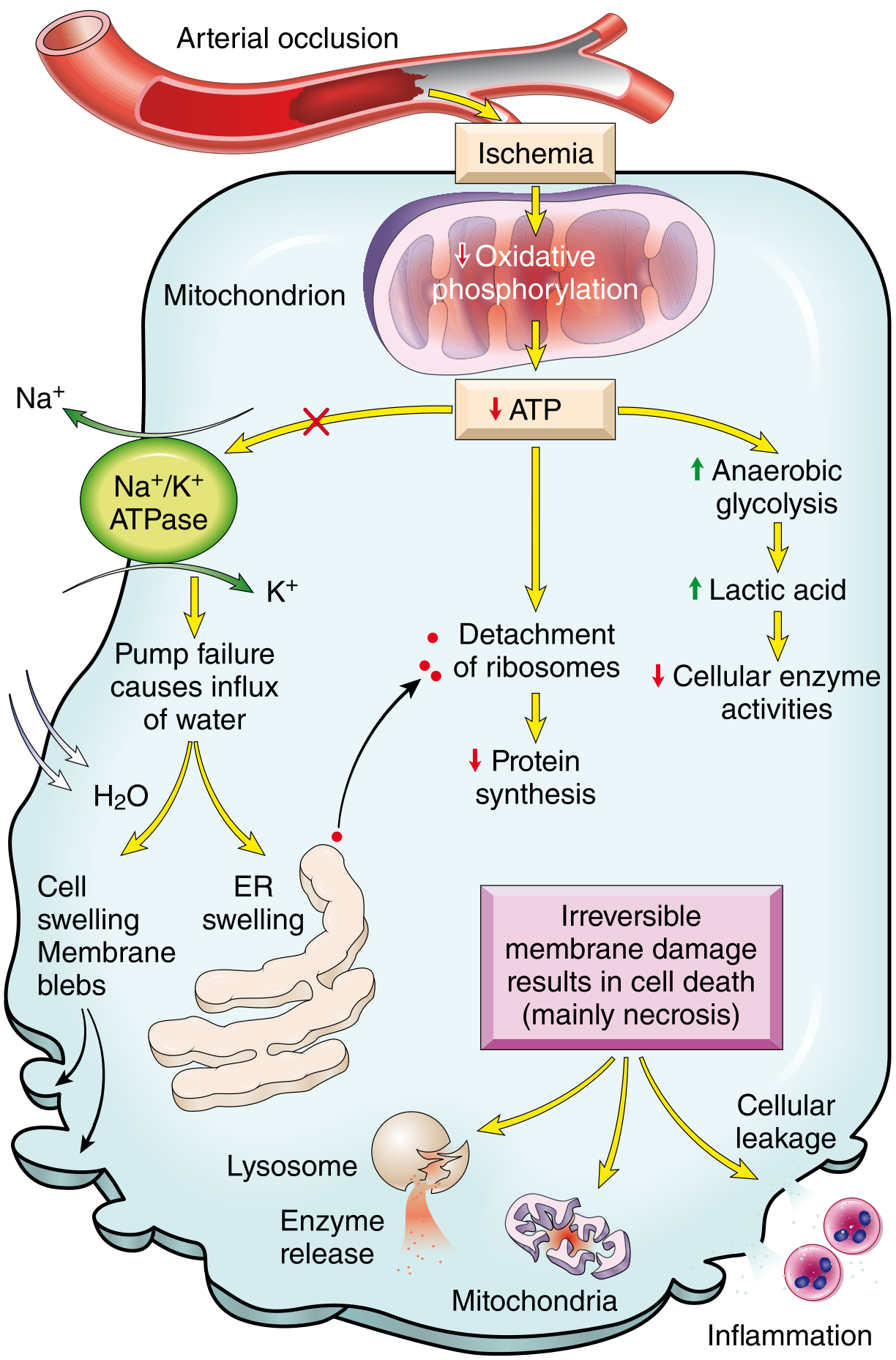
Fig. 2.24 - Functional and morphologic consequences of decreased intracellular ATP in ischemic cell injury. Arterial occlusion → ischemia → failed oxidative phosphorylation → ATP depletion → cascade of cell injury events.
The core sequence following ischemia is:
Step 1 - ATP Depletion
- Oxygen deprivation → failure of oxidative phosphorylation in mitochondria → ATP drops
- Cell switches to anaerobic glycolysis → lactic acid accumulates → intracellular pH falls → enzymes are inhibited
Step 2 - Failure of the Na⁺/K⁺ ATPase pump (Reversible Injury)
- ATP depletion disables the Na⁺/K⁺ pump
- Na⁺ and water flood into the cell → cellular swelling and membrane blebs
- K⁺ leaks out
- ER swells → ribosomes detach → reduced protein synthesis
Step 3 - Calcium Influx (Transition to Irreversible Injury)
- Intracellular Ca²⁺ rises due to failure of Ca²⁺-ATPase pumps and increased membrane permeability
- Ca²⁺ activates phospholipases (membrane damage), proteases (cytoskeletal breakdown), endonucleases (DNA damage), and ATPases (further ATP depletion)
- Ca²⁺ overload opens the mitochondrial permeability transition (MPT) pore → further ATP depletion and cytochrome c release → apoptosis
Step 4 - Irreversible Injury (Necrosis)
- Severe mitochondrial swelling and vacuolation
- Lysosome rupture → enzyme release → autodigestion
- Plasma membrane disruption → cellular leakage of contents → sterile inflammation
- Death by coagulative necrosis (most tissues) or liquefactive necrosis (brain)
Summary of Reversible vs. Irreversible Ischemic Injury
| Feature | Reversible (Mild/Early) | Irreversible (Severe/Prolonged) |
|---|---|---|
| Mitochondria | Swollen, small densities | Amorphous densities, vacuolation |
| Cell membrane | Blebbing | Rupture |
| Lysosomes | Intact | Ruptured |
| Nuclear changes | None | Pyknosis, karyolysis, karyorrhexis |
| Outcome | Recovery if flow restored | Necrosis/apoptosis |
Ischemia-Reperfusion Injury
Paradoxically, restoring blood flow to ischemic tissue can worsen the injury by activating new damaging processes. This is called ischemia-reperfusion (I/R) injury, and it is clinically important in myocardial infarction treated with thrombolysis/PCI and in stroke treated with thrombolytics.
Mechanisms of Reperfusion Injury
1. Oxidative stress (ROS burst)
- Reoxygenation generates a sudden surge of reactive oxygen species (ROS) - from leukocytes, damaged endothelium, and parenchymal cells
- Antioxidant defenses depleted during ischemia → cells are particularly susceptible
2. Intracellular calcium overload
- Ca²⁺ influx worsens during reperfusion (membrane damage + ROS injury to the sarcoplasmic reticulum)
- Opens mitochondrial permeability transition (MPT) pore → ATP depletion → cell death
3. Inflammation
- Ischemic cells release "danger signals" and cytokines → recruitment of neutrophils into reperfused tissue
- Neutrophils cause additional injury via ROS, proteases, and eicosanoids
- Upregulation of adhesion molecules on hypoxic endothelium amplifies neutrophil recruitment
4. Complement activation
- IgM antibodies deposit in ischemic tissues
- On reperfusion, complement cascade is activated → further cell injury and inflammation
Infarction - The End Result
An infarct is an area of ischemic necrosis caused by occlusion of either the arterial supply or venous drainage. Most infarcts result from arterial thrombosis or embolism.
Types of Infarcts
| Type | Appearance | Occurs in |
|---|---|---|
| White (pale) infarct | Pale, firm, wedge-shaped | Solid organs with end-arteries: heart, spleen, kidney |
| Red (hemorrhagic) infarct | Red, congested | Organs with dual blood supply (lung, liver) or after venous occlusion; loose spongy tissues |
- Both types are wedge-shaped, with the occluded vessel at the apex and the organ surface at the base
- Early margins are indistinct and slightly hemorrhagic; later margins become better defined by a rim of hyperemia (inflammation)
- Most infarcts are replaced by scar tissue (fibrosis) over time
- Exception: Brain infarcts undergo liquefactive necrosis, not coagulative necrosis
Factors Influencing Infarct Development
| Factor | Details |
|---|---|
| Vascular anatomy | Dual blood supplies (liver, lung, forearm) protect against infarction; end-arterial circulations (kidney, spleen, heart) do not |
| Rate of occlusion | Slow occlusion (e.g., gradual atherosclerosis) allows development of collateral circulation, reducing infarct size |
| Tissue vulnerability to hypoxia | Neurons: irreversible injury after 3-4 minutes; Myocardium: after 20-30 minutes; Fibroblasts: survive for hours |
Tissue Vulnerability: Time Windows
| Tissue | Time to Irreversible Injury |
|---|---|
| Neurons (CNS) | 3-4 minutes |
| Myocardial cells | 20-30 minutes |
| Renal tubular cells | ~60 minutes |
| Fibroblasts / connective tissue | Hours |
This is why time-to-treatment is so critical in stroke ("time is brain") and myocardial infarction ("time is muscle").
Protective Response: HIF-1
Cells respond to ischemia by inducing Hypoxia-Inducible Factor-1 (HIF-1), a transcription factor that:
- Promotes angiogenesis (new vessel formation)
- Stimulates cell survival pathways
- Enhances anaerobic glycolysis
Investigational compounds that promote HIF-1 signaling are in development. The most established protective strategy for ischemic brain/spinal cord injury is therapeutic hypothermia - lowering core body temperature reduces metabolic demands, decreases cell swelling, suppresses ROS formation, and inhibits inflammation.
Clinical Correlations
| Organ | Ischemic Syndrome |
|---|---|
| Heart | Angina pectoris, Myocardial infarction (MI) |
| Brain | TIA, Ischemic stroke |
| Kidney | Renal infarction, Acute tubular necrosis (ATN) |
| Intestine | Mesenteric ischemia, bowel infarction |
| Limbs | Peripheral arterial disease, critical limb ischemia |
| Lung | Pulmonary infarction (usually from embolism) |
Sources: Robbins & Kumar Basic Pathology (9780323790185); Robbins, Cotran & Kumar Pathologic Basis of Disease (9780443264528)
Shock
Finding Sources
Searching PubMed
shock pathophysiology management
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
Reading File
Finding Sources
Reading File
Here is a thorough overview of Shock, drawn primarily from Robbins & Kumar Basic Pathology.
Shock
Shock is a state in which diminished cardiac output or reduced effective circulating blood volume impairs tissue perfusion, leading to cellular hypoxia. At the outset, cellular injury is reversible. Prolonged shock, however, leads to irreversible tissue injury and is frequently fatal.
Types of Shock
| Type | Clinical Examples | Principal Mechanism |
|---|---|---|
| Cardiogenic | Myocardial infarction, ventricular rupture, arrhythmia, cardiac tamponade, pulmonary embolism | Failure of the myocardial pump - intrinsic damage, extrinsic compression, or outflow obstruction |
| Hypovolemic | Hemorrhage, severe burns, vomiting/diarrhea | Inadequate blood or plasma volume → low cardiac output |
| Septic | Gram-positive/negative bacteremia, fungal sepsis, toxic shock syndrome | Peripheral vasodilation, blood pooling, endothelial activation/injury, DIC, cytokine cascades |
| Neurogenic | Anesthesia, spinal cord injury | Loss of vascular tone → vasodilation |
| Anaphylactic | IgE-mediated hypersensitivity (drugs, bee stings, foods) | Systemic vasodilation + increased vascular permeability |
Stages of Shock
Shock evolves through three progressive stages (best documented in hypovolemic shock, but common to all types):
Stage 1 - Nonprogressive (Compensated)
The body activates neurohumoral compensatory mechanisms to maintain blood pressure and vital organ perfusion:
- Baroreceptor reflexes → sympathetic activation
- Catecholamine release → tachycardia, peripheral vasoconstriction
- ADH release → renal water retention
- Renin-angiotensin-aldosterone (RAA) activation → fluid conservation
- Net effect: Blood is redistributed away from skin/gut toward heart and brain
- Clinical signs: Tachycardia, cool/clammy/pale skin (except septic shock, which causes warm flushed skin due to vasodilation)
Stage 2 - Progressive
When the underlying cause is not corrected:
- Widespread tissue hypoxia → anaerobic glycolysis → lactic acid accumulation
- Lactic acidosis blunts arteriolar vasomotor response → arterioles dilate → blood pools in microcirculation
- Peripheral pooling → decreased cardiac output + endothelial ischemia → risk of DIC
- Vital organs begin to fail
Stage 3 - Irreversible
Even if hemodynamic defects are corrected, survival is not possible:
- Lysosomal enzyme leakage → worsens cell injury
- Myocardial contractility deteriorates severely
- Gut ischemia → intestinal flora enter circulation → superimposed bacteremic shock
- Renal failure from ischemic kidney injury
- Death follows multiorgan failure
Pathogenesis of Septic Shock (Detailed)
Septic shock is the most mechanistically complex type. It is most commonly triggered by gram-positive bacteria, followed by gram-negative bacteria and fungi, and carries a mortality of 20-30% even with modern care.
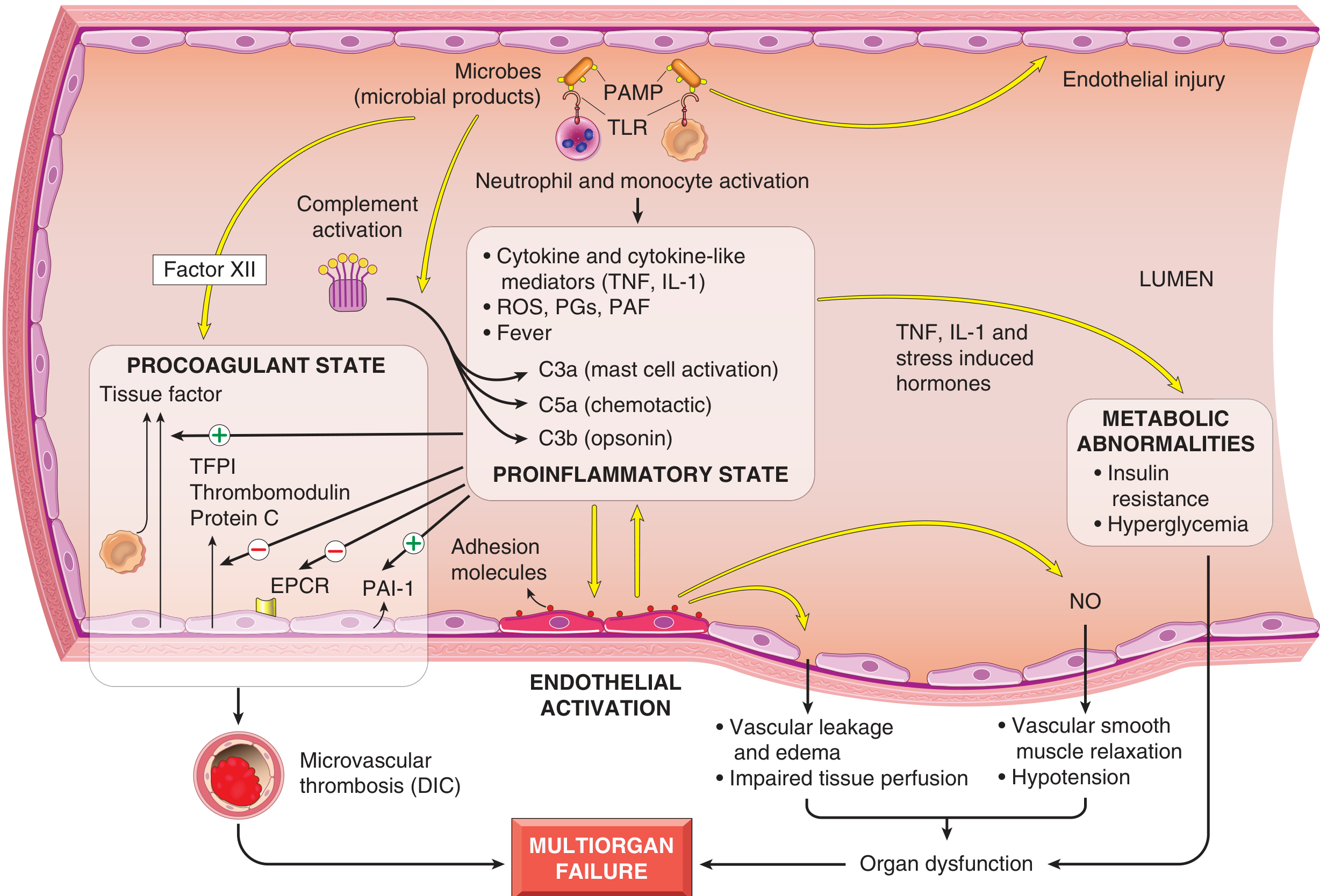
FIG. 3.19 - Major pathogenic pathways in septic shock. Microbial products activate endothelial cells and innate immune elements, triggering a cascade leading to procoagulant state, proinflammatory state, metabolic abnormalities, and ultimately multiorgan failure.
Four Core Pathogenic Mechanisms:
1. Proinflammatory Response
- Microbial PAMPs (pathogen-associated molecular patterns) activate:
- Toll-like receptors (TLRs) on macrophages and neutrophils
- G-protein-coupled receptors (detect bacterial peptides)
- C-type lectin receptors (dectins - detect fungi)
- Activated cells release: TNF, IL-1, IL-12, IL-18, ROS, prostaglandins, PAF
- Complement cascade activation → C3a (mast cell activation), C5a (chemotaxis), C3b (opsonin)
- With time, a counterregulatory immunosuppressive phase follows, worsening vulnerability to secondary infections
2. Endothelial Activation and Injury
- Cytokines loosen endothelial tight junctions → vascular leakage + widespread protein-rich edema
- Edema impairs nutrient delivery and waste removal
- Activated endothelium upregulates nitric oxide (NO) and vasoactive mediators → vascular smooth muscle relaxation → systemic hypotension
- Upregulated adhesion molecules amplify leukocyte recruitment
3. Procoagulant State - DIC
- Sepsis alters coagulation balance:
- Increased: Tissue factor production by monocytes/endothelium, PAI-1 (blocks fibrinolysis)
- Decreased: TFPI, thrombomodulin, endothelial protein C receptor (all anticoagulants)
- Result: Systemic thrombin activation → fibrin-rich microvascular thrombi throughout the body
- Full DIC occurs in up to 50% of septic patients → consumption of clotting factors and platelets → paradoxical concurrent bleeding and thrombosis
4. Metabolic Abnormalities
- Cytokines (TNF, IL-1) + stress hormones → insulin resistance + hyperglycemia
- Enhanced gluconeogenesis; suppressed insulin release
- Late phase: Adrenal insufficiency (functional or from adrenal necrosis - Waterhouse-Friderichsen syndrome)
- Mitochondrial damage + cellular hypoxia → lactic acidosis
Superantigens (e.g., toxic shock syndrome toxin) are a special case: polyclonal T-cell activators that cause massive cytokine release, producing a shock syndrome clinically similar to septic shock.
Morphologic Effects of Shock
The pathophysiologic effects are essentially those of hypoxic injury, caused by a combination of hypoperfusion and microvascular thrombosis.
| Organ | Morphologic Change |
|---|---|
| Kidney | Acute tubular necrosis (ATN); fibrin thrombi in glomeruli |
| Adrenal glands | Cortical lipid depletion (increased steroid synthesis under stress) |
| Lung | "Shock lung" - diffuse alveolar damage (especially in septic/traumatic shock); lungs are relatively resistant in pure hypovolemic shock |
| Brain | Ischemic encephalopathy |
| Heart | Subendocardial hemorrhage and necrosis |
| GI tract | Ischemic enterocolitis; mucosal hemorrhage and ulceration |
| Liver | Centrilobular necrosis ("nutmeg" pattern) |
Except for neurons and cardiomyocytes, affected tissues may recover completely if the patient survives.
Clinical Features
| Feature | Hypovolemic / Cardiogenic | Septic |
|---|---|---|
| Blood pressure | Low | Low (may initially be maintained) |
| Pulse | Weak, rapid | Rapid |
| Skin | Cool, clammy, pale/cyanotic | Initially warm, flushed (vasodilation) → later cool |
| Breathing | Rapid | Rapid |
| Urine output | Oliguria | Oliguria (late) |
As shock progresses, worsening renal function dominates: oliguria, acidosis, and electrolyte imbalances.
Prognosis
| Type of Shock | Mortality (with appropriate treatment) |
|---|---|
| Hypovolemic (young, healthy patient) | <10% |
| Septic shock | 20-30% |
| Cardiogenic shock | 50-80% |
The severity and outcome of septic shock depend on: extent and virulence of infection, immune status of the host, comorbidities, and the pattern of mediator production. The complexity of overlapping pathways explains why targeting single mediators (e.g., anti-TNF antibodies) has largely failed in clinical trials.
Sources: Robbins & Kumar Basic Pathology (9780323790185); Robbins, Cotran & Kumar Pathologic Basis of Disease (9780443264528); Harrison's Principles of Internal Medicine 22E (9781265977061)
This is a shared conversation. Sign in to Orris to start your own chat.