Amyloidosis mbbs 2nd year patho
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Finding Sources
I have all the content needed from Robbins Cotran. Now let me compile the comprehensive MBBS 2nd year pathology notes on Amyloidosis:
Amyloidosis - Pathology Notes (MBBS 2nd Year)
Source: Robbins, Cotran & Kumar - Pathologic Basis of Disease
Definition
Amyloidosis is a group of disorders characterized by extracellular deposition of abnormal protein aggregates (amyloid) in various tissues and organs, causing progressive tissue damage and functional compromise.
- The term "amyloid" comes from "amylose" (starch) because early chemists mistook the deposits for starch due to their similar staining behavior - but amyloid is actually a protein, not a carbohydrate.
Physical Nature of Amyloid
- By electron microscopy: continuous, non-branching fibrils, ~8-10 nm in diameter
- Each fibril consists of stacks of protofilaments with a beta (β)-pleated sheet conformation
- This β-pleated sheet structure is shared by ALL types of amyloid and is responsible for the characteristic Congo red staining
Chemical Nature of Amyloid
Approximately 95% fibril proteins + 5% serum amyloid P (SAP) component and glycoproteins.
Three Major Biochemical Forms:
| Type | Fibril Protein | Precursor Protein | Clinical Setting |
|---|---|---|---|
| AL (Amyloid Light chain) | Immunoglobulin light chains (predominantly λ) | Plasma cell dyscrasias (myeloma) | Primary amyloidosis |
| AA (Amyloid-Associated) | AA protein (non-Ig fragment) | Serum amyloid A (SAA) - an acute phase reactant | Secondary/reactive amyloidosis |
| ATTR (Transthyretin) | Transthyretin (TTR) | Wild-type TTR or mutant TTR | Senile systemic or hereditary amyloidosis |
Other types:
- Aβ2M (Beta-2 microglobulin): dialysis-associated amyloidosis; deposits in joints/tendons
- Aβ (A-beta peptide): Alzheimer disease; amyloid in cerebral plaques and vessels
- AIAPP (Islet amyloid polypeptide): Type 2 diabetes; deposits in islets of Langerhans
- AE (Endocrine amyloid): medullary carcinoma of thyroid (procalcitonin)
Pathogenesis (Mechanism of Amyloid Formation)
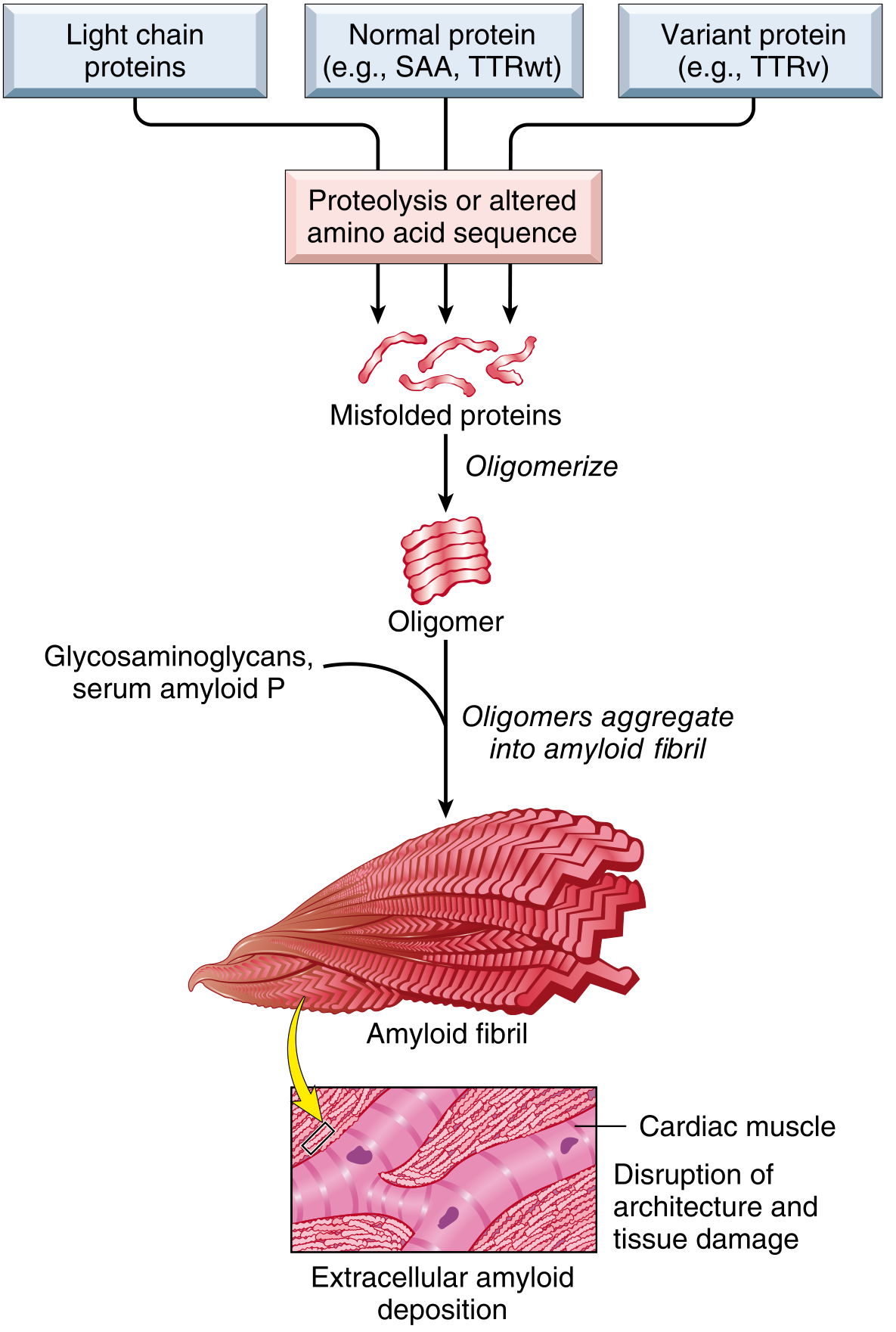
Fig 6.44 - Robbins Cotran: Misfolded proteins oligomerize and aggregate into amyloid fibrils that deposit extracellularly, disrupting tissue architecture
Three mechanisms lead to amyloid formation:
- Overproduction of a normal protein that has an intrinsic tendency to fold improperly (e.g., overproduction of immunoglobulin light chains in myeloma)
- Acquired mutations leading to protein misfolding (e.g., mutant TTR in hereditary amyloidosis)
- Normal aging changes in protein configuration (e.g., wild-type TTR in senile amyloidosis)
In all cases: Misfolded protein → Oligomers → Amyloid fibrils → Extracellular deposition + association with glycosaminoglycans (heparan sulfate, dermatan sulfate) and serum amyloid P component.
Classification of Amyloidosis
1. Systemic (Generalized) Amyloidosis
| Type | Also Called | Amyloid | Cause |
|---|---|---|---|
| Primary | AL amyloidosis | AL | Plasma cell dyscrasia (multiple myeloma, MGUS) |
| Secondary (Reactive) | AA amyloidosis | AA | Chronic inflammatory diseases (TB, osteomyelitis, RA, Crohn's, bronchiectasis) |
| Hemodialysis-associated | Aβ2M amyloidosis | Aβ2M | Renal failure on long-term dialysis |
| Hereditary (Familial) | ATTR, etc. | Variable | Autosomal dominant mutations |
2. Localized Amyloidosis
- Senile cardiac amyloidosis (wild-type TTR) - elderly men
- Senile cerebrovascular amyloidosis (Alzheimer disease)
- Endocrine tumors (medullary carcinoma of thyroid, islet cell tumors)
- Isolated amyloid deposits in aging heart, lung, skin
Morphology (Gross & Microscopic)
Gross Appearance
- Affected organ: enlarged, gray, waxy, firm consistency
Microscopic (H&E):
- Amyloid appears as amorphous, eosinophilic, hyaline, extracellular substance
- Deposition is always extracellular, begins adjacent to basement membranes
- Progressively encroaches on and destroys cells
- Perivascular and vascular deposits are common in AL type
Special Stains - KEY EXAM POINTS:
| Stain | Finding |
|---|---|
| Congo red (ordinary light) | Pink/red deposits |
| Congo red (polarized light) | Apple-green birefringence ✦ (pathognomonic) |
| Crystal violet / Methyl violet | Metachromasia (red-violet deposits stain purple) |
| Thioflavin T/S | Yellow-green fluorescence under UV |
| PAS | Positive (due to associated glycoproteins) |
The apple-green birefringence with Congo red under polarized light is caused by the cross-β-pleated sheet configuration of amyloid fibrils - it is the gold standard histologic diagnostic test.
Organ-Specific Changes
Kidney (Most common & serious)
- Most frequently affected organ
- Deposits in glomeruli (mesangial matrix first, then GBM), also interstitium, vessels
- Glomerular: subtle mesangial thickening → capillary narrowing → eventual obliteration ("flooded glomerulus")
- Clinically: Nephrotic syndrome → progressive renal failure
Spleen
- Sago spleen: deposits limited to splenic follicles → tapioca-like granules on gross section
- Lardaceous spleen: deposits in sinusoidal walls and red pulp → large "map-like" areas → waxy, translucent, lard-like appearance
Liver
- Deposits along sinusoids (space of Disse) and portal areas
- Kupffer cells and hepatocytes compressed → hepatomegaly
- Liver function usually preserved until late
Heart
- Deposits in myocardial interstitium and conduction system
- Restrictive cardiomyopathy → heart failure, arrhythmias
- Characteristic "sparkling" appearance on echocardiography
Tongue
- Macroglossia - characteristic of AL/primary amyloidosis
Peripheral Nerves
- Carpal tunnel syndrome (especially in Aβ2M amyloidosis)
- Peripheral neuropathy
Adrenals, Thyroid, Pituitary
- Often involved in secondary amyloidosis
Clinical Features
Primary (AL) Amyloidosis:
- Associated with multiple myeloma or monoclonal gammopathy
- Heart, GI tract, nerves, skin, tongue predominantly affected
- Macroglossia (virtually pathognomonic when present)
- Periorbital purpura ("raccoon eyes") - from vascular fragility
Secondary (AA) Amyloidosis:
- Complicates chronic infections (TB, osteomyelitis, bronchiectasis), rheumatoid arthritis, Crohn's disease, leprosy
- Kidneys, liver, spleen, adrenals, lymph nodes predominantly affected
- Nephrotic syndrome is the dominant clinical presentation
Hereditary (ATTR) Amyloidosis:
- Mutations in transthyretin (TTR) gene
- Most common: V30M mutation (valine→methionine at position 30)
- Peripheral neuropathy, autonomic neuropathy, cardiomyopathy
Diagnosis
| Method | Use |
|---|---|
| Biopsy + Congo red stain | Gold standard |
| Abdominal fat pad aspiration | Least invasive, most commonly used first |
| Rectal biopsy | High yield for systemic amyloidosis |
| Renal biopsy | If nephrotic syndrome present |
| Bone marrow biopsy | If plasma cell dyscrasia suspected |
| Serum/urine protein electrophoresis (SPEP/UPEP) | Detect monoclonal protein in AL |
| Immunohistochemistry | Typing AA vs AL vs ATTR |
| Mass spectrometry | Definitive typing, gold standard for typing |
| SAP scintigraphy | Distribution of amyloid deposits (research tool) |
Prognosis
- Generally poor - no curative treatment available
- Depends heavily on type, extent of organ involvement
- AL amyloidosis: median survival ~1-2 years without treatment
- AA amyloidosis: treating the underlying disease can halt progression
- Newer targeted therapies for ATTR amyloidosis (tafamidis, patisiran) are now available
High-Yield Summary for Exams
| Feature | Key Point |
|---|---|
| Nature of amyloid | Misfolded proteins with β-pleated sheet |
| Stain used | Congo red → apple-green birefringence under polarized light |
| AL amyloid | Associated with plasma cell dyscrasia (myeloma) |
| AA amyloid | Reactive/secondary to chronic inflammation |
| Most common organ | Kidney (nephrotic syndrome) |
| Macroglossia | AL (primary) amyloidosis |
| Sago spleen | Follicular deposits (white pulp) |
| Lardaceous spleen | Red pulp / sinusoidal deposits |
| Dialysis amyloid | β2-microglobulin type |
| Alzheimer's amyloid | Aβ (beta-amyloid) - cerebral plaques |
| Medullary thyroid carcinoma | Amyloid from procalcitonin |
Source: Robbins, Cotran & Kumar - Pathologic Basis of Disease (9780443264528), Chapter 6
This is a shared conversation. Sign in to Orris to start your own chat.