Myosarcoma in a child
Reading File
Finding Sources
Searching PubMed
rhabdomyosarcoma children
Searching the Web
rhabdomyosarcoma children 2025 treatment guidelines
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Loading Image
I now have comprehensive material to write a thorough answer. Let me compile everything.
Rhabdomyosarcoma (RMS) in Children
Rhabdomyosarcoma is a malignant mesenchymal tumor with skeletal muscle differentiation and is the most common soft tissue sarcoma of childhood. It accounts for 3-4.5% of all pediatric malignancies, and in the head/neck region it is the dominant soft tissue sarcoma.
Subtypes
Four histologic subtypes are recognized. The first two are predominantly pediatric:
| Subtype | Frequency | Age | Key Features |
|---|---|---|---|
| Embryonal | ~50% | Children | Soft, gray, infiltrative; spindle/round cells in myxoid stroma; rhabdomyoblasts with strap-like cytoplasm and cross-striations |
| Alveolar | ~20% | Children/adolescents | Firmer; round cells in alveolar pattern; PAX3/PAX7-FOXO1 fusions; worse prognosis |
| Pleomorphic | ~20% | Adults | Large multinucleate bizarre cells |
| Spindle cell/sclerosing | ~10% | All ages | Fusiform cells in fascicles, storiform pattern |
Sarcoma botryoides is a variant of embryonal RMS that grows as polypoid masses in hollow viscera (urinary bladder, vagina) - classically in young girls.
Histology
Fig. 19.44 (Robbins & Kumar Basic Pathology) - Rhabdomyosarcoma:
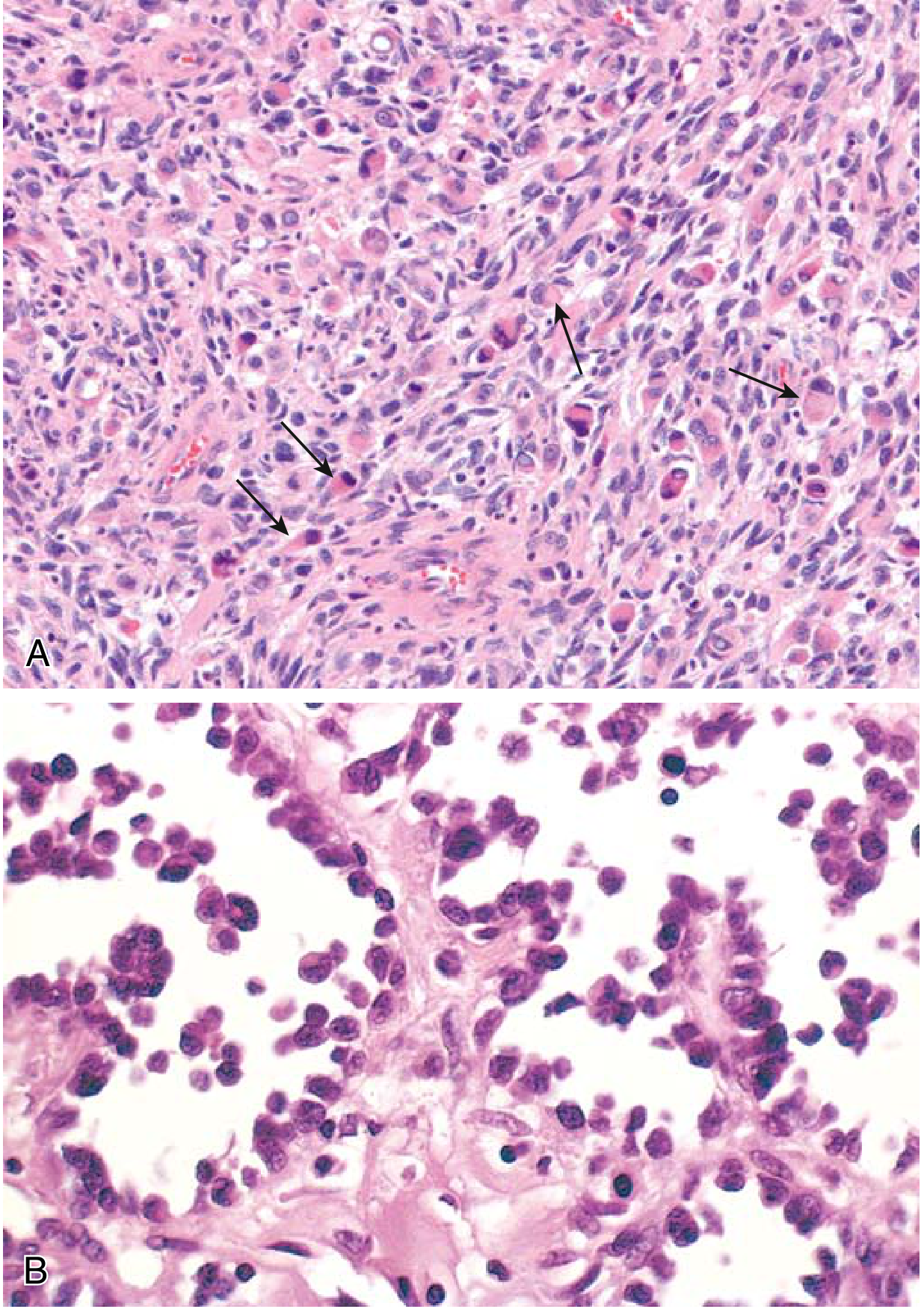
(A) Embryonal subtype - malignant cells from primitive/round to eosinophilic with skeletal muscle differentiation (arrows). (B) Alveolar subtype - spaces lined by dyscohesive, uniform round tumor cells.
Fig. 204.3 (Cummings Otolaryngology) - Embryonal RMS:
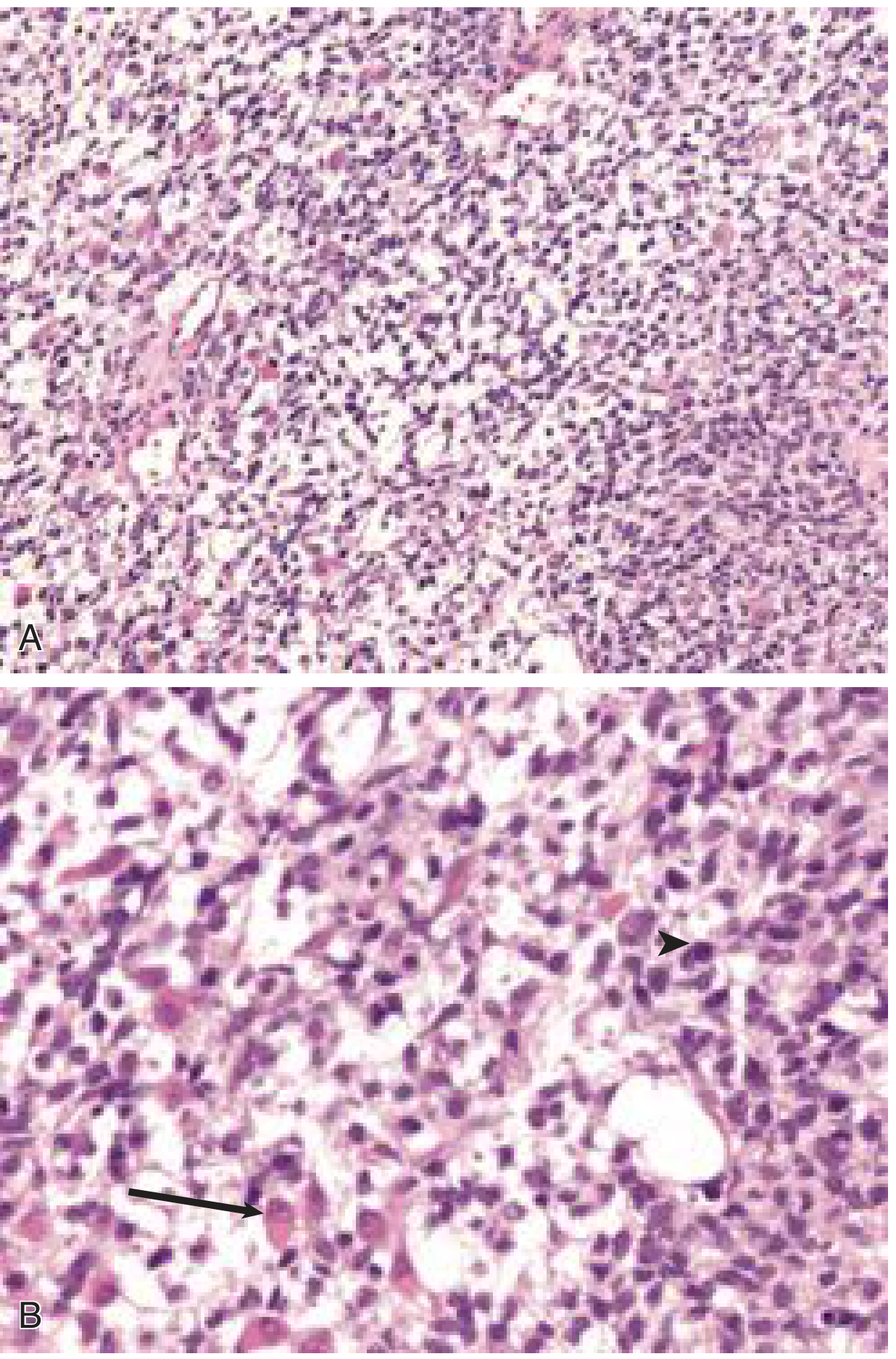
(A) Stellate and spindle cells in a myxoid background. (B) Higher magnification showing a rhabdomyoblastic cell with eccentric nucleus and eosinophilic cytoplasm (arrow) and a mitotic figure (arrowhead).
Genetics / Molecular Biology
- Alveolar RMS: Characterized by translocations t(2;13) or t(1;13), fusing PAX3 or PAX7 to FOXO1 (previously known as FKHR). PAX3 normally initiates skeletal muscle differentiation; the chimeric fusion protein interferes with differentiation and drives oncogenesis - a mechanism analogous to transcription factor fusions in acute leukemia. PAX3-FOXO1 fusions confer a worse prognosis.
- Embryonal/Pleomorphic RMS: Genetically heterogeneous; no consistent single translocation.
Common Sites in Children
Pediatric RMS often arises in locations that do not normally contain much skeletal muscle, which supports origin from undifferentiated mesenchymal stem cells:
- Head and neck (~35%): Orbit (most common single subsite, ~25% of H&N RMS), parameningeal sites (middle ear/mastoid, nasal cavity, paranasal sinuses, parapharyngeal space, pterygopalatine/infratemporal fossa)
- Genitourinary tract: Bladder, prostate, vagina, paratesticular
- Extremities: Particularly alveolar subtype
Parameningeal tumors are close to the skull base and carry higher risk of intracranial extension.
Presentation
- Firm, often asymptomatic mass (most common)
- Site-specific symptoms: nasal obstruction, rhinorrhea, epistaxis, otitis media with aural polyp, proptosis (orbital), hematuria/urinary obstruction (GU), cranial nerve deficits (parameningeal)
- Cranial nerve deficits suggest skull base erosion
Diagnosis and Workup
- Biopsy (open or core): The definitive step
- IHC: Muscle-specific proteins - myogenin (most specific), MyoD1, desmin, muscle-specific actin; cross-striations on H&E may be visible
- Imaging:
- MRI: Isointense to muscle on T1; high signal on T2; best for soft tissue extent and perineural spread
- CT: Better for bony erosion (skull base assessment)
- FDG-PET/CT: Sensitive for macroscopic metastatic disease
- Technetium-99 bone scan: For osseous metastases
- Staging workup: CBC, electrolytes, renal/liver function, coagulation; bone marrow aspirate and CSF cytology (for parameningeal/metastatic staging)
Staging
Two complementary systems are used together by the Children's Oncology Group (COG):
1. Pretreatment TNM Staging (Table 204.6)
| Stage | Site | Tumor | Size | Nodes | Mets |
|---|---|---|---|---|---|
| 1 | Favorable (orbit, non-parameningeal H&N) | T1 or T2 | Any | Any N | M0 |
| 2 | Unfavorable | T1 or T2 | ≤5 cm | N0/NX | M0 |
| 3 | Unfavorable | T1 or T2 | >5 cm or N1 | Any N | M0 |
| 4 | Any | T1 or T2 | Any | Any | M1 |
2. COG Surgical-Pathologic Group (IRS Clinical Group)
| Group | Definition |
|---|---|
| I | Localized, completely resected, clear margins, no nodal disease |
| II | Completely removed but with microscopic residual OR involved regional nodes grossly removed |
| III | Incomplete removal - gross residual disease after biopsy or subtotal resection |
| IV | Distant metastases at diagnosis |
These two systems, combined with histologic subtype, determine risk stratification (low, intermediate, high) which drives treatment intensity.
Treatment
Management is multimodal and guided by COG protocols. Pediatric patients should be enrolled in clinical trials when available.
Surgery
- Complete surgical resection with clear margins is the goal when achievable without major functional morbidity
- Mutilating operations are avoided - chemotherapy and radiation are given first to reduce tumor size (neoadjuvant approach) for unresectable disease
- Accurate pathologic group assignment after surgery is critical
Chemotherapy
- The backbone regimen is VAC (Vincristine, Actinomycin D, Cyclophosphamide) - established through decades of Intergroup Rhabdomyosarcoma Study Group (IRSG) trials
- Higher-risk disease uses more intensive regimens
Radiation Therapy
- Used for groups II-IV and selected group I alveolar tumors
- Orbital RMS: Radiation as part of multimodal therapy (avoids enucleation)
- Recent 2025 meta-analysis data (Fukushima et al., PMID 40494941) compared proton beam therapy vs. photon radiotherapy - proton therapy is increasingly preferred in children to spare surrounding tissues
- For metastatic disease: Conditional recommendation for whole lung irradiation (WLI) at 15 Gy/10 fractions for pulmonary metastases (improves EFS but consistent OS benefit not proven); SABR 30-60 Gy/3-5 fractions for limited lung lesions
Prognosis
- In the 1960s, 5-year OS was ~50%; with multimodal therapy it rose to ~70% by the 1990s and continues to improve
- Favorable prognosis: Orbital location, embryonal histology, localized disease (Group I/II), Stage 1
- Unfavorable prognosis: Alveolar histology (PAX3-FOXO1 especially), parameningeal location, metastatic disease (Group IV, Stage 4) - 5-year OS ~25% for metastatic disease
- The botryoid variant of embryonal RMS carries a particularly good prognosis
Relapsed Disease
At least 1/3 of localized RMS and 60-70% of metastatic RMS experience progressive disease or relapse. The 2025 European pediatric soft tissue sarcoma study group (EpSSG) published treatment recommendations for relapsed RMS, and a 2026 systematic review (PMID 42093305) examines response outcomes as survival predictors in relapsed/refractory pediatric RMS.
Sources: Robbins & Kumar Basic Pathology, p. 802 | Cummings Otolaryngology Head and Neck Surgery, pp. 3853-3856 | Campbell's Operative Orthopaedics 15th Ed 2026, p. 1278 | Fukushima et al. 2025, Int J Clin Oncol, PMID 40494941 | Defachelles et al. 2025, Br J Cancer, PMID 40913059
This is a shared conversation. Sign in to Orris to start your own chat.