Myocardial infarction
Reading File
Finding Sources
Finding Sources
Searching PubMed
"myocardial infarction"[MeSH Terms] AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive, well-sourced content. Let me compile the full response.
Myocardial Infarction (MI)
Definition
Myocardial infarction is necrosis of heart muscle resulting from ischemia. The 2018 Fourth Universal Definition (joint U.S./European task force) defines MI as "the presence of acute myocardial injury detected by abnormal cardiac biomarkers in the setting of evidence of acute myocardial ischemia." - Robbins & Kumar Basic Pathology, p. 354
Epidemiology
In the United States, approximately 605,000 patients experience a new AMI and 200,000 a recurrent AMI each year. About half of all AMI-related deaths occur before reaching the hospital. In-hospital mortality has declined from ~10% to ~5% in the era of primary PCI. The 1-year mortality rate after AMI is approximately 15%, and is ~fourfold higher in patients >75 years old. - Harrison's Principles of Internal Medicine 22E, p. 2160
Men are at greater risk than women, but the gap narrows with age. Women are relatively protected during reproductive years; menopause (with declining estrogen) is associated with exacerbation of coronary artery disease. Approximately 10% of MIs occur before age 40, and 45% before age 65. - Robbins & Kumar Basic Pathology, p. 354
Classification (Fourth Universal Definition - 5 Types)
| Type | Mechanism |
|---|---|
| Type 1 | Spontaneous MI due to atherosclerotic plaque disruption/erosion with thrombosis |
| Type 2 | MI due to supply-demand mismatch (e.g., coronary spasm, anemia, tachyarrhythmia) |
| Type 3 | MI causing sudden cardiac death before biomarkers can be drawn |
| Type 4a/4b | PCI-related MI or in-stent thrombosis |
| Type 5 | CABG-related MI |
- Sabiston Textbook of Surgery, p. 3071
STEMI vs. NSTEMI
- STEMI (ST-Elevation MI): Complete occlusion; transmural injury; ST elevation on ECG
- NSTEMI (Non-ST-Elevation MI): Partial/transient occlusion; subendocardial injury; no ST elevation but troponin elevated
- Unstable angina: Plaque disruption + thrombus, but no biomarker rise; increasingly treated aggressively
Pathophysiology
Coronary Artery Occlusion - Step by Step
STEMI typically occurs when an atherosclerotic plaque undergoes sudden disruption or erosion, triggering a cascade: - Harrison's Principles, p. 2160
- Atheromatous plaque eroded or disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces - exposing subendothelial collagen and necrotic plaque contents
- Platelets adhere, aggregate, and release thromboxane A2, ADP, and serotonin - promoting further aggregation and vasospasm
- Coagulation cascade activated by tissue factor exposure - thrombin converts fibrinogen to fibrin
- Glycoprotein IIb/IIIa receptors on platelets develop high affinity for fibrinogen - cross-linking platelets
- Within minutes, the thrombus may completely occlude the coronary lumen
- Angiography within 4 hours of MI onset demonstrates coronary thrombosis in ~90% of cases
Plaques prone to disruption characteristically have: rich lipid core, thin fibrous cap, expansive remodeling, neovascularization, and plaque hemorrhage. - Harrison's p. 2160
Cellular Response to Ischemia
- Within seconds: aerobic metabolism ceases; ATP drops; potentially noxious metabolites (lactic acid) accumulate
- Within minutes: loss of contractility (reversible at this stage)
- After 20-40 minutes of persistent ischemia: irreversible damage and coagulative necrosis
- Earliest detectable necrosis: sarcolemmal membrane disruption - intracellular macromolecules leak into circulation
- If blood flow restored before irreversible injury: myocardium preserved ("stunned myocardium" - transiently noncontractile for days)
- In 80-90% of cardiac deaths: cause is ventricular fibrillation from myocardial irritability, not pump failure
Guyton & Hall Medical Physiology notes that cardiac muscle requires ~1.3 mL O2/100g/min just to survive; normal resting delivery is ~8 mL O2/100g/min. With even 15-30% of normal resting coronary flow, muscle will not die - but in the central zone of a large infarct with near-zero collateral flow, muscle dies.
Subendocardial Vulnerability
Subendocardial muscle is especially susceptible because:
- It is the last area to receive blood from epicardial vessels
- It is exposed to relatively high intramural pressures that impede blood inflow
- It has higher oxygen consumption
Thus any condition compromising coronary flow causes damage first in the subendocardium, spreading outward (wavefront phenomenon). - Guyton & Hall, p. 271; Robbins p. 355
Coronary Artery Territories and Infarct Patterns
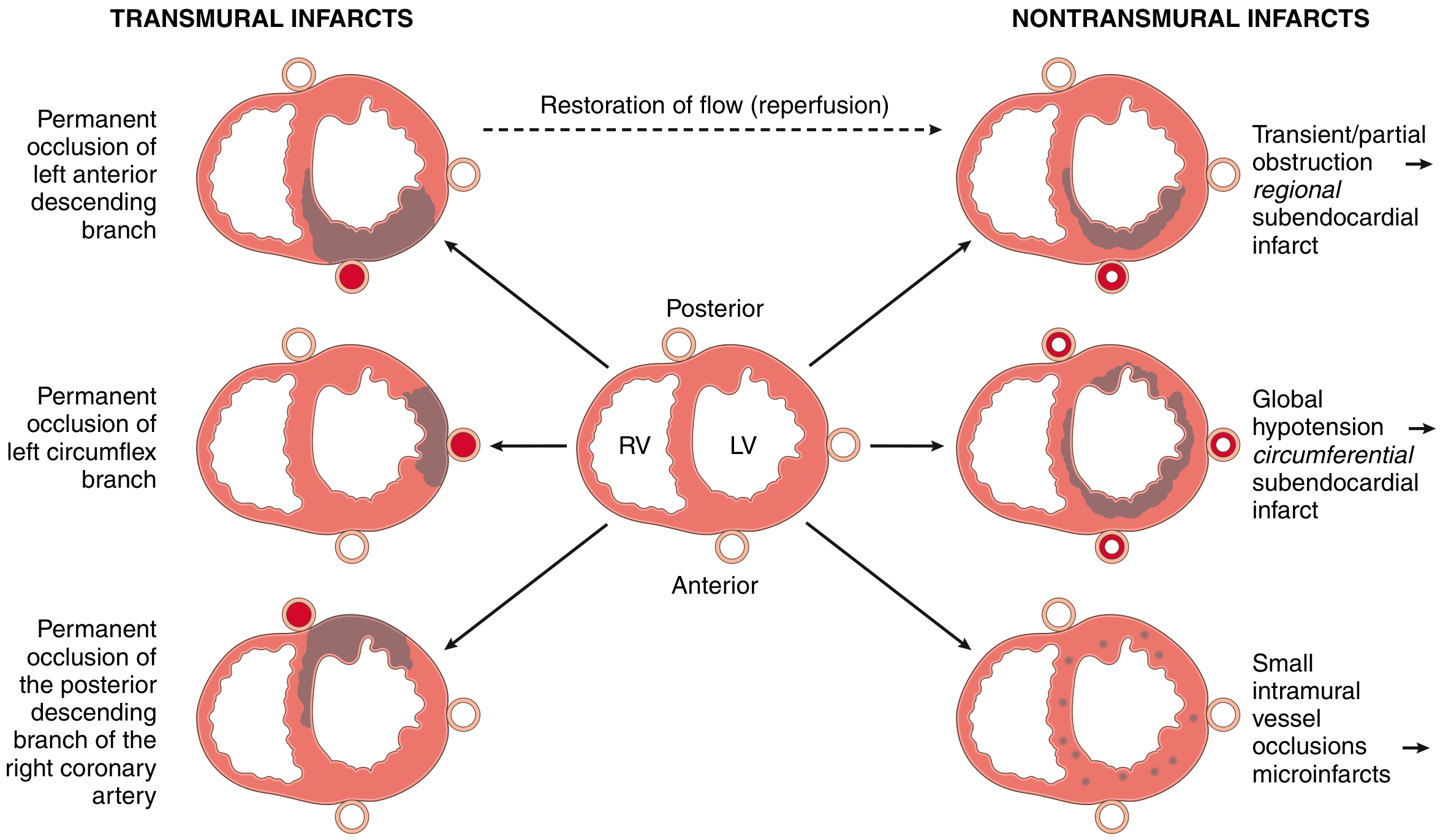
| Artery | Territory | % of MIs |
|---|---|---|
| Proximal LAD | Anterior LV wall, anterior 2/3 of septum, apex | 40-50% |
| Proximal LCX | Lateral LV wall | 15-20% |
| Proximal RCA | Right ventricle, posterior/inferior LV (right dominant) | 30-40% |
- Right dominant circulation (90% of individuals): RCA gives rise to posterior descending artery
- 15-30% of posterior/posteroseptal MIs also extend into the right ventricle
- Isolated RV infarction: only 1-3% of IHD cases
- Robbins & Kumar Basic Pathology, p. 356
Morphological Timeline
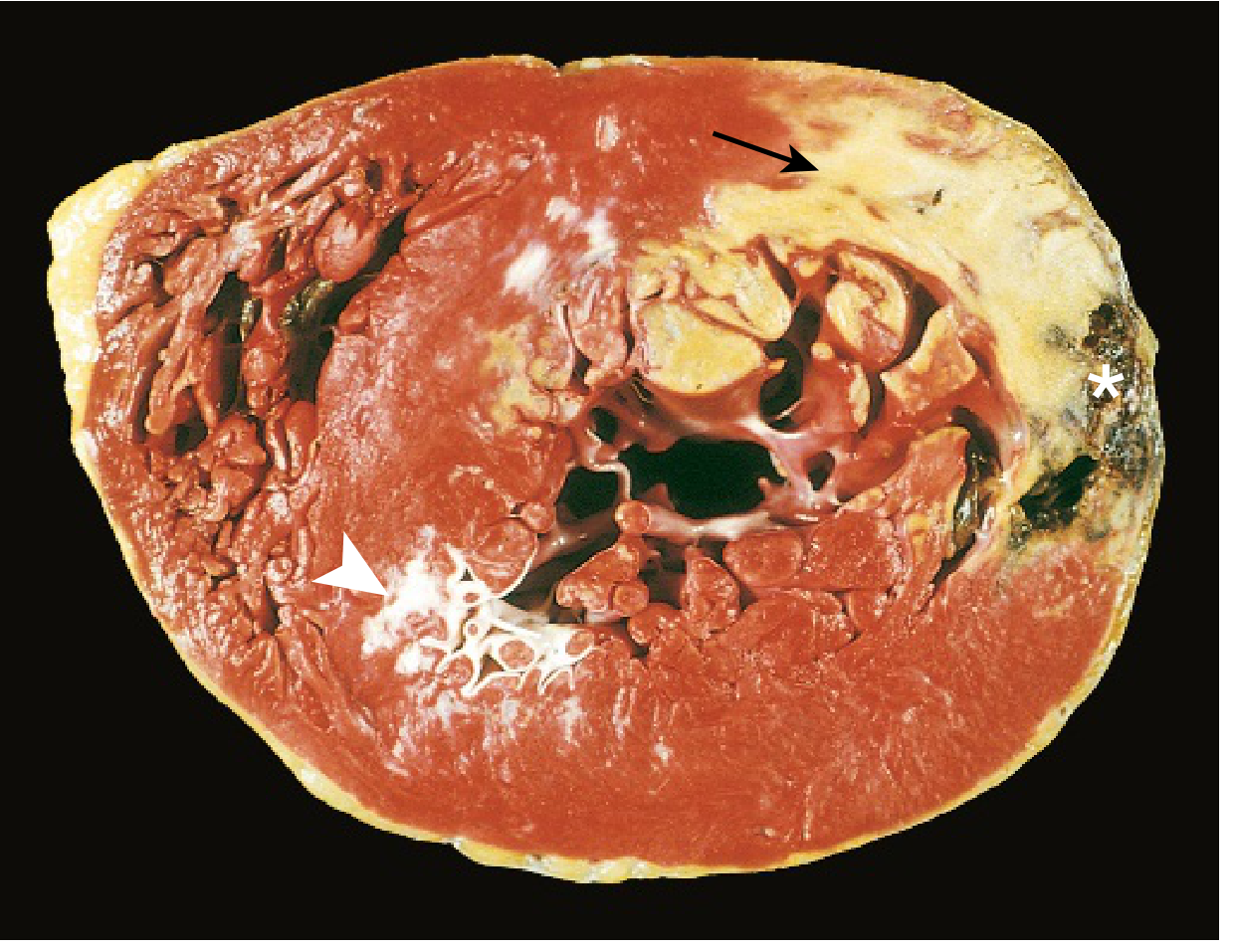
| Time Frame | Gross Features | Light Microscopy |
|---|---|---|
| 0-30 min | None | None (reversible) |
| 30 min - 4 hrs | None | Usually none; variable waviness at border; sarcolemmal disruption |
| 4-12 hrs | Occasionally dark mottling | Onset coagulation necrosis; edema; hemorrhage |
| 12-24 hrs | Dark mottling | Coagulation necrosis; pyknosis of nuclei; hypereosinophilic myocytes; early neutrophils |
| 1-3 days | Mottling + yellow-tan center | Coagulation necrosis; loss of nuclei/striations; increased neutrophils |
| 3-7 days | Hyperemic border; yellow-tan softening | Disintegration of dead myofibers; early macrophage phagocytosis at border |
| 7-10 days | Maximally yellow-tan and soft; depressed red-tan margins | Well-developed phagocytosis; early granulation tissue at margins |
| 10-14 days | Red-gray depressed borders | Established granulation tissue; new vessels; collagen deposition |
| 2-8 weeks | Gray-white scar, progressing inward | Increasing collagen; decreasing cellularity |
| >2 months | Scarring complete | Dense collagenous scar |
- Robbins & Kumar Basic Pathology, Table 9.2, p. 357
Key diagnostic point: Infarcts <12 hours old are usually NOT grossly apparent. TTC (triphenyl tetrazolium chloride) staining is used to visualize infarcts >3 hours old - necrotic areas appear pale (enzyme leaks out), healthy myocardium stains red-brick.
ECG Changes
The three major ECG abnormalities in acute MI (Ganong's physiology): - Ganong's Review of Medical Physiology 26e, p. 534
| Defect | Current Flow | ECG Change |
|---|---|---|
| Rapid repolarization | Out of infarct | ST elevation |
| Decreased resting membrane potential (K+ loss) | Into infarct | TQ depression (manifested as ST elevation) |
| Delayed depolarization | Out of infarct | ST elevation |
- Acute STEMI: ST elevation in leads overlying the infarct; reciprocal ST depression in opposite leads
- Evolving: After days-weeks, ST changes subside; dead muscle electrically silent
- Old MI: Q waves (pathological) appear in relevant leads ("Q-wave MI"); reflects lack of normal depolarization vector
- Non-Q-wave MI (NSTEMI): Tends to be less transmural but carries high risk of early reinfarction
Clinical Presentation
Symptoms
- Chest pain: Heavy, crushing, pressure-like; substernal; radiates to left arm, jaw, neck, back
- Duration: >20-30 minutes (unlike angina)
- Associated: Diaphoresis, nausea/vomiting, dyspnea, sense of impending doom
- Atypical presentations (especially in women, elderly, diabetics): Epigastric pain, fatigue, syncope, silent MI
Signs
- Anxiety, diaphoresis, pallor
- Tachycardia or bradycardia (especially inferior MI - vagal)
- S3 or S4 gallop
- New murmur (papillary muscle dysfunction, VSD)
- Pulmonary crackles (if LV failure)
- Hypotension (cardiogenic shock)
Biomarkers
| Marker | Rise | Peak | Return to Normal | Notes |
|---|---|---|---|---|
| cTnI / cTnT | 2-4 hrs | 24-48 hrs (STEMI) | 7-10 days (cTnI); 10-14 days (cTnT) | Preferred marker; most specific |
| CK-MB | 4-6 hrs | 18-24 hrs | 48-72 hrs | Useful for reinfarction detection |
| Myoglobin | 1-2 hrs | 6-9 hrs | 24 hrs | First to rise; not cardiac-specific |
High-sensitivity troponin assays (hs-cTn) are the current standard. A rise and/or fall pattern with at least one value above the 99th percentile is required for diagnosis. - Harrison's p. 2162
Early reperfusion causes earlier peaking of biomarkers (rapid washout from infarct zone).
Non-specific inflammatory markers: WBC 12,000-15,000/μL (peaks 2-3 days), elevated ESR, CRP
Causes of Death After AMI
- Decreased cardiac output - "systolic stretch" (ischemic zone bulges during systole instead of contracting - reduces net pumping efficiency) - Guyton & Hall, p. 271
- Pulmonary edema - damming of blood in pulmonary vessels
- Ventricular fibrillation - most common cause; majority of VF deaths within first 24h (half in first hour)
- Cardiac rupture (rare but catastrophic) - usually ventricular free wall, papillary muscles, or interventricular septum at 3-7 days
Complications
| Complication | Timing | Notes |
|---|---|---|
| Arrhythmias (VF, VT, AF, heart block) | Immediate | VF most deadly; heart block common in inferior MI (RCA) |
| Cardiogenic shock | Hours-days | Pump failure; ~10% of STEMI; high mortality |
| Acute MR | Acute/subacute | Papillary muscle ischemia/rupture |
| Ventricular septal rupture | 3-7 days | New harsh murmur; surgical emergency |
| Free wall rupture | 3-7 days | Hemopericardium; tamponade; usually fatal |
| Pericarditis (early) | 1-3 days | Pericardial friction rub; pleuritic chest pain |
| Dressler syndrome | 2-10 weeks | Autoimmune; fever, pericarditis, pleuritis |
| Mural thrombus | Days-weeks | LV thrombus over akinetic segment; embolic risk |
| LV aneurysm | Weeks-months | Persistent ST elevation; paradoxical wall motion |
| Heart failure | Chronic | Infarct scar; ventricular remodeling |
Management
Prehospital
- Immediate aspirin (if no contraindication)
- Rapid transport to PCI-capable center
- Defibrillator available (most out-of-hospital deaths = VF in first hour)
- Prehospital fibrinolysis in remote settings if ECG-confirmed STEMI and trained personnel
Emergency Department - Initial Medications
"MONA" + dual antiplatelet + anticoagulation:
- Morphine (IV for pain, reduces sympathetic drive)
- Oxygen (if SpO2 <90%)
- Nitroglycerin (IV if BP >90 mmHg systolic; avoid in RV infarction and phosphodiesterase inhibitor use)
- Aspirin (162-325 mg, chewed, immediately) - Tintinalli's EM
- P2Y12 inhibitor (clopidogrel, ticagrelor, or prasugrel)
- Anticoagulation (unfractionated heparin, LMWH, or bivalirudin)
- Beta-blockers (oral; avoid if cardiogenic shock risk, acute decompensation, or significant bradycardia)
- ACE inhibitors (begin within 24h if no hypotension/renal failure - reduce LV remodeling)
- Statins (high-intensity: atorvastatin 40-80 mg)
Reperfusion Strategy
Primary PCI is the preferred strategy when available within 120 minutes of first medical contact:
- Superior to fibrinolysis in reducing mortality, reinfarction, and stroke
- Door-to-balloon time target: <90 minutes
- Greatest benefit in: age <75, no prior MI, anterior STEMI
Fibrinolysis when PCI unavailable within 120 min:
- Acceptable within 12 hours of symptom onset (most benefit within 3 hours)
- Agents: Alteplase (tPA), reteplase, tenecteplase (TNK)
- Contraindications: recent surgery/stroke, active bleeding, hypertensive emergency
Cardiogenic Shock Management
(Tintinalli's Emergency Medicine, p. 397-398)
| Drug | Dose | Indication |
|---|---|---|
| Dobutamine | 2-20 mcg/kg/min | Inotrope of choice if SBP ≥90 mmHg |
| Norepinephrine | 2 mcg/min, titrate | Preferred vasopressor if SBP <70 mmHg |
| Dopamine | 3-50 mcg/kg/min | Inotrope + vasopressor (increased dysrhythmia risk) |
| Milrinone | 0.5 mcg/kg/min | Inotrope if on beta-blocker |
- Mechanical circulatory support (IABP, Impella) as bridge to revascularization
- Emergency PCI or CABG for definitive treatment of cardiogenic shock
Secondary Prevention (Post-MI)
- Dual antiplatelet therapy (aspirin + P2Y12) for 12 months
- Beta-blockers (for at least 3 years post-MI; recent 2025 meta-analysis questions benefit in preserved EF without HF - PMID 39298680)
- ACE inhibitor or ARB (especially if EF reduced or DM)
- Aldosterone antagonist (if EF <40% + DM or HF)
- High-intensity statin (target LDL-C <55 mg/dL)
- Cardiac rehabilitation, lifestyle modification, smoking cessation
Recent Evidence Update
A 2025 meta-analysis (PMID 39298680, European Journal of Preventive Cardiology) suggests that beta-blockers for secondary prevention after MI in patients without reduced ejection fraction or heart failure may not provide mortality benefit - this represents an evolving area that may shift future guidelines.
A 2024 meta-analysis (PMID 39378245, European Heart Journal) on sudden cardiac death after MI using pooled individual participant data provides updated risk stratification data relevant to ICD prophylaxis decisions.
Sources: Robbins & Kumar Basic Pathology (9780323790185) | Harrison's Principles of Internal Medicine 22E (9781265977061) | Guyton & Hall Textbook of Medical Physiology (9780443111013) | Ganong's Review of Medical Physiology 26e (9781260122404) | Tintinalli's Emergency Medicine (9781260019933) | Sabiston Textbook of Surgery (9780443124341)
This is a shared conversation. Sign in to Orris to start your own chat.