Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Reading File
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND "pathophysiology"[MeSH Terms]
Searching Images
ARDS acute respiratory distress syndrome pathophysiology mechanism diagram
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND "lung injury"[MeSH Terms]
Finding Sources
Finding Sources
Searching PubMed
"ARDS" AND "neutrophil" AND "alveolar damage"
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the information needed to deliver a comprehensive, well-structured answer. Here it is:
Mechanism of Acute Respiratory Distress Syndrome (ARDS)
ARDS is a syndrome of noncardiogenic pulmonary edema caused by increased alveolar-capillary permeability, not elevated hydrostatic pressure. The result is protein-rich exudate flooding the alveoli, profound hypoxemia (PaO₂/FiO₂ < 300 by Berlin criteria), and decreased respiratory system compliance.
Precipitating Insults
Injury reaches the lung either directly or indirectly:
| Direct (Pulmonary) | Indirect (Systemic) |
|---|---|
| Pneumonia (bacterial, viral, COVID-19) | Sepsis (most common) |
| Aspiration of gastric contents | Major trauma |
| Toxic inhalation | Pancreatitis |
| Pulmonary contusion | Multiple blood transfusions |
| Near-drowning | Drug overdose / adverse medication effect |
Core Pathophysiology: The Alveolar-Capillary Barrier
The central event is disruption of the alveolar-capillary membrane, which requires injury to both:
- Pulmonary microvascular endothelium — loss of endothelial barrier integrity is both necessary and sufficient for ARDS to develop.
- Alveolar epithelium — damage here is considered the key precipitating event; it disrupts barrier integrity AND prevents alveolar fluid clearance.
Mechanisms of cell death include necrosis, apoptosis, coagulation activation, and mechanical stretch. — Murray & Nadel's Textbook of Respiratory Medicine
Step-by-Step Pathogenesis
1. Initial Lung Injury & Macrophage Activation
A direct or systemic insult activates alveolar macrophages, which release proinflammatory cytokines and chemokines — especially TNF-α, IL-1β, IL-6, and IL-8 (CXCL8). These act as powerful neutrophil chemoattractants and amplify the inflammatory cascade through NF-κB–mediated transcription.
2. Neutrophil Sequestration and Transmigration
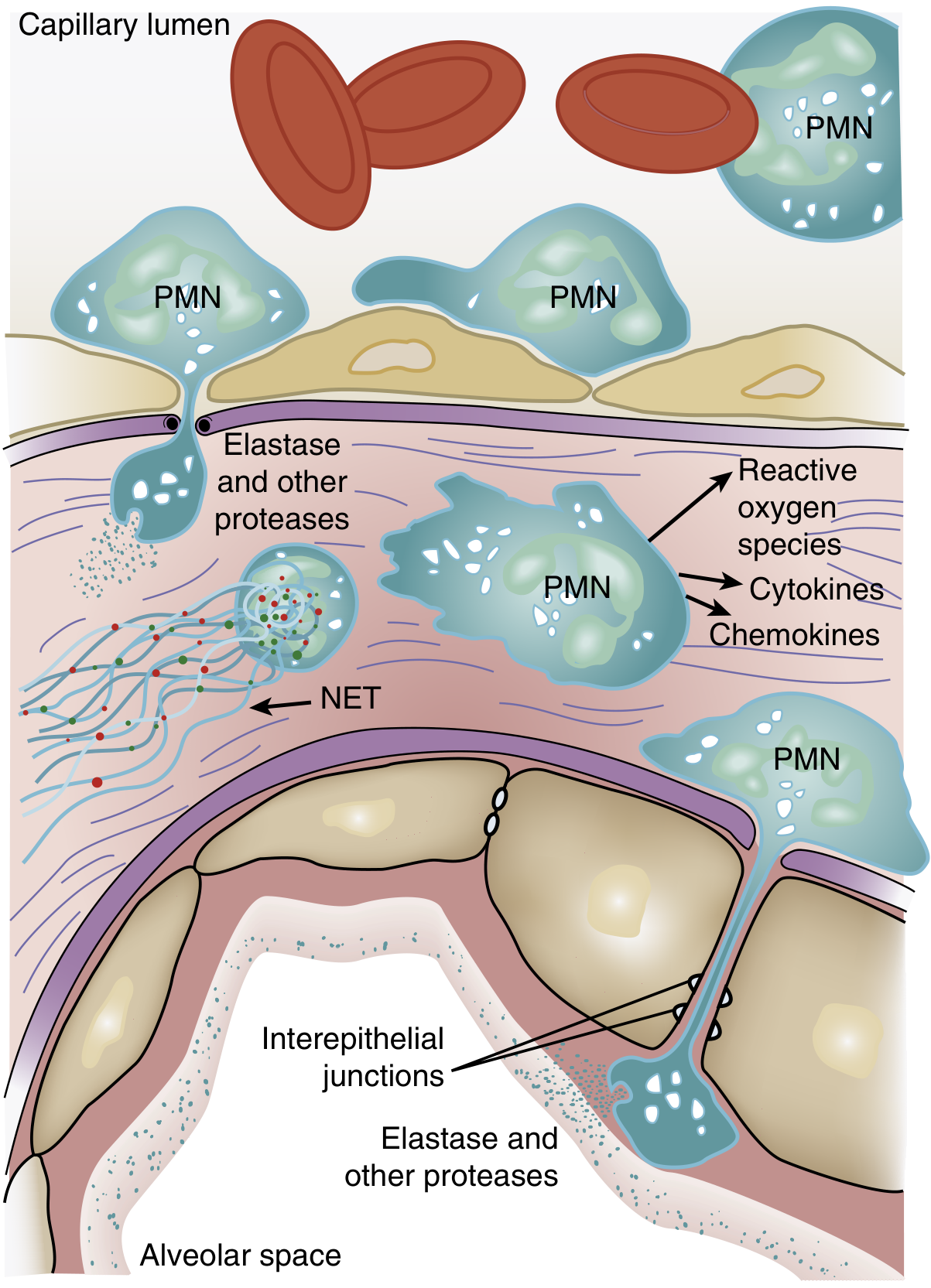
One of the earliest signs of ARDS — even before hypoxemia — is transient leukopenia caused by neutrophil sequestration in the pulmonary microvasculature. Because the average pulmonary capillary diameter is smaller than the average neutrophil diameter, neutrophils must deform to pass through. Activated neutrophils become mechanically "stiff" via actin cytoskeletal changes and cannot negotiate narrow capillary segments, leading to trapping.
Once sequestered, neutrophils transmigrate into the interstitium — initially without requiring adhesion molecules such as L-selectin or β2-integrins — and then migrate into the alveolar space. Sequestered neutrophils also induce endothelial barrier breakdown, perpetuating their own migration.
3. Neutrophil-Mediated Damage
Activated neutrophils deploy a cytotoxic arsenal:
| Weapon | Effect |
|---|---|
| Reactive oxygen species (ROS) | Oxidative membrane injury to epithelial and endothelial cells |
| Neutrophil elastase (NE) | Degrades cadherins in adherens junctions → alveolar flooding; degrades surfactant protein A; cleaves growth factors and cytokines |
| Metalloproteinases | Extracellular matrix degradation, barrier disruption |
| Defensins and cationic peptides | Direct cellular cytotoxicity |
| TNF-α, IL-1β (released by PMNs) | Amplify the inflammatory response |
| Neutrophil extracellular traps (NETs) | Web-like DNA-histone-antimicrobial peptide structures that trap pathogens but also cause endothelial damage and thrombus formation |
NE degradation of epithelial and endothelial E-cadherin (the main component of adherens junctions) is particularly destructive, as it directly predisposes to alveolar flooding. — Murray & Nadel's Textbook of Respiratory Medicine
4. Cytokine Storm and Amplification
Proinflammatory cytokines — especially TNF-α, IL-1β, IL-6, IL-8 — are released in large quantities from activated macrophages, neutrophils, and injured epithelial cells. Key signal transduction pathways include:
- p38 MAP kinase: activated by LPS; stimulates TNF-α production and macrophage inflammatory protein-2 (MIP-2), amplifying neutrophil chemotaxis
- PI3-kinase-γ: preferentially activated in neutrophils by IL-8 and bacterial peptides; knockout mice show markedly decreased neutrophil accumulation and cytokine production after endotoxin
- NF-κB: major transcriptional driver of the proinflammatory program
This creates a self-amplifying inflammatory loop in which each wave of neutrophil-mediated damage generates more cytokines, which recruit more neutrophils.
5. Surfactant Dysfunction
Neutrophil elastase degrades surfactant protein A — this has been confirmed in BAL fluid from ARDS patients. Phospholipase A₂ (elevated in pancreatitis-associated ARDS) enzymatically degrades surfactant directly. Loss of surfactant function leads to alveolar instability and collapse, worsening ventilation-perfusion mismatch. (Unlike neonatal RDS where surfactant deficiency is the primary driver, its role in adult ARDS is secondary and contributory, as four large RCTs of exogenous surfactant in adults failed to show mortality benefit.)
6. Coagulation Abnormalities
Plasma extravasation into the airspaces activates the coagulation cascade via surfactant- and macrophage-derived procoagulants. This leads to intra-alveolar fibrin deposition (forming hyaline membranes) and intravascular fibrin deposition, contributing to pulmonary hypertension. Fibrin and fibrinogen fragments are themselves chemotactic for neutrophils, perpetuating inflammation.
7. Impaired Alveolar Fluid Clearance
Normally, sodium is reabsorbed across the alveolar epithelium via apical Na⁺ channels → basolateral Na⁺/K⁺-ATPase, creating an osmotic gradient that clears edema. In ARDS this mechanism is severely impaired by:
- Hypoxia → downregulates epithelial Na⁺ channel expression and Na⁺/K⁺-ATPase activity
- Apoptosis/necrosis of type I and II alveolar cells
- Proinflammatory cytokines and oxidants → defects in transcellular ion transport
Protein clearance from flooded alveoli proceeds at only 1–2%/hour (vs. 10–20%/hour for fluid), resulting in progressive protein concentration in the alveolar space. — Fishman's Pulmonary Diseases and Disorders
8. Angiopoietin-2 and Vascular Instability
Angiopoietin-2 (Ang-2) is an endogenous antagonist of Ang-1 and a potent destabilizer of vascular integrity. Ang-2 is released by activated endothelium and is markedly elevated in ARDS. It acts on Tie-2 receptors to promote endothelial permeability, leukocyte adhesion, and inflammation. Plasma Ang-2 levels correlate with ARDS severity and mortality.
Three Phases of Pathology (Diffuse Alveolar Damage)
Though ~50% of ARDS patients demonstrate histologic DAD, it remains the classic pathological correlate:
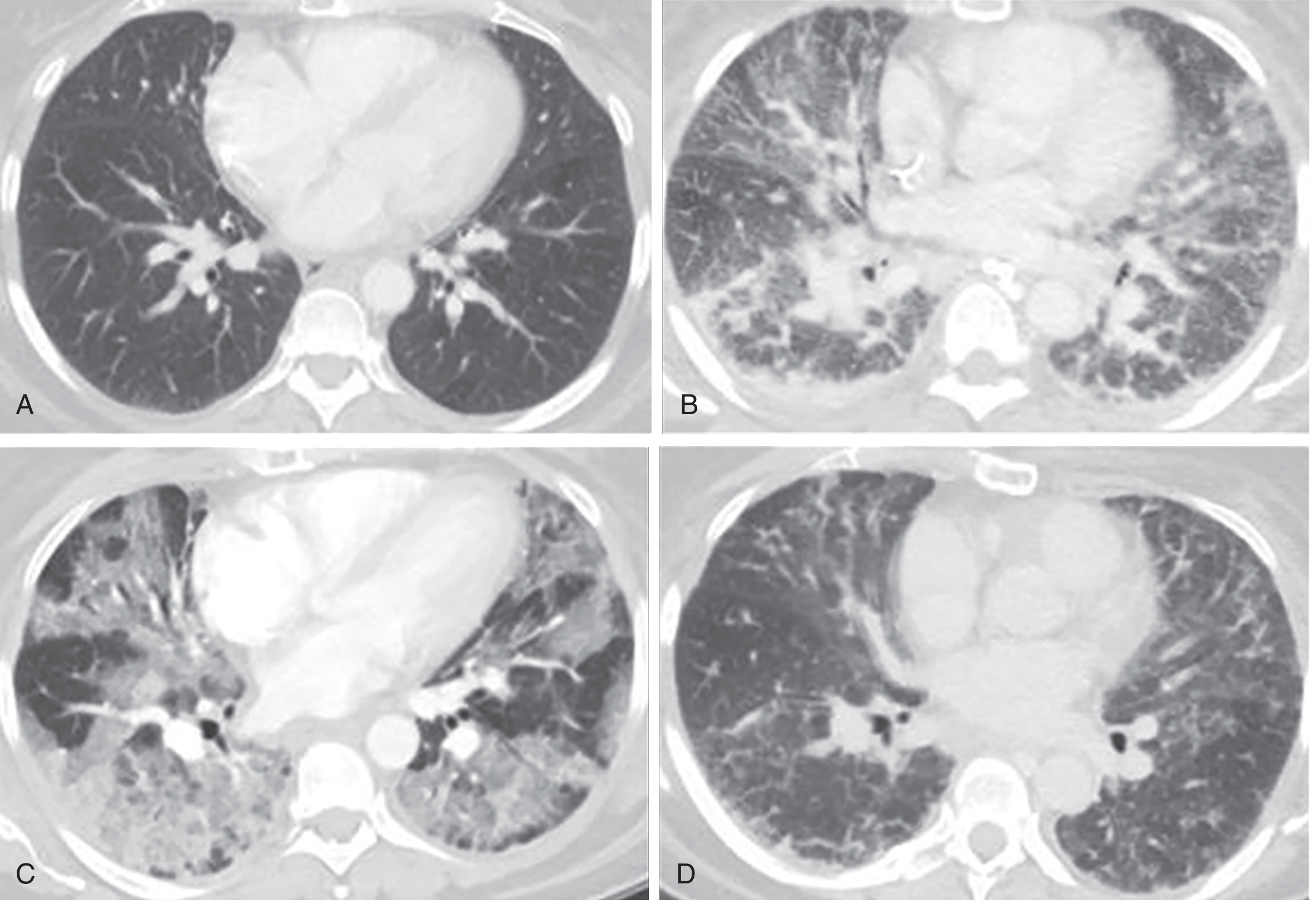
| Phase | Timing | Pathology |
|---|---|---|
| Exudative | Days 1–7 | Protein-rich alveolar edema, hyaline membrane formation (cellular debris + fibrin + surfactant components), diffuse neutrophil infiltration, widespread epithelial disruption |
| Proliferative | Days 7–21 | Hyaline membrane reorganization, fibrosis begins, obliteration of pulmonary capillaries, interstitial/alveolar collagen deposition, neutrophil numbers decline |
| Fibrotic | >2 weeks | Pulmonary fibrosis in a subset; fibroproliferation may actually begin simultaneously with, not after, the exudative phase — N-terminal procollagen III peptide is detectable in BAL fluid as early as 24 hours after onset |
Physiological Consequences
| Consequence | Mechanism |
|---|---|
| Severe hypoxemia | Alveolar flooding → intrapulmonary right-to-left shunt |
| Decreased compliance | Alveolar filling + surfactant loss + edema stiffening |
| Increased dead space | Vascular occlusion → elevated VD/VT, requiring high minute ventilation |
| Pulmonary hypertension | Hypoxic vasoconstriction + intravascular fibrin + compression by positive-pressure ventilation |
Summary
ARDS is not a single-mechanism disease. It begins with an inciting insult that activates macrophages and triggers a cytokine storm → neutrophil sequestration and transmigration → release of ROS, elastases, NETs, and further cytokines → alveolar-capillary barrier disruption → protein-rich exudate flooding the alveoli → surfactant inactivation, fibrin deposition (hyaline membranes), and impaired edema clearance → severe hypoxemia, low compliance, and dead-space physiology. The cycle is self-amplifying and in severe cases progresses to fibroproliferation and irreversible fibrosis.
Sources: Murray & Nadel's Textbook of Respiratory Medicine (Chapter 134), Fishman's Pulmonary Diseases and Disorders (Chapter 140), Goldman-Cecil Medicine (Chapter 91), Sabiston Textbook of Surgery (Chapter 11)
This is a shared conversation. Sign in to Orris to start your own chat.