Iron deficiency anemia vs thalassemia
Reading File
Finding Sources
Finding Sources
Searching PubMed
"iron deficiency anemia" AND "thalassemia" AND "differential diagnosis"
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive data to write a thorough comparison. Let me compile the full answer.
Iron Deficiency Anemia vs. Thalassemia
Both are microcytic, hypochromic anemias - this shared morphology is the central diagnostic challenge. Below is a thorough comparison.
1. Pathophysiology
Iron Deficiency Anemia (IDA)
IDA is the most common nutritional disorder worldwide. Hemoglobin synthesis fails because iron - the core component of heme - is depleted. The sequence of depletion is:
- Storage iron (ferritin/hemosiderin) depleted first
- Then transport iron (serum iron falls, TIBC rises, transferrin saturation drops)
- Finally, erythropoiesis becomes iron-restricted → microcytic, hypochromic RBCs
Iron balance is normally maintained by duodenal absorption regulated by hepcidin. In IDA, hepcidin falls (removing its brake on ferroportin), increasing intestinal iron uptake. Despite this compensatory response, supply still cannot meet demand. - Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 612-614
Thalassemia
Thalassemia is a genetically heterogeneous disorder caused by germline mutations that reduce synthesis of either α-globin (α-thalassemia, chromosome 16) or β-globin (β-thalassemia, chromosome 11). Anemia arises via two mechanisms:
- Decreased hemoglobin content → underhemaglobinized, microcytic red cells
- Imbalance of globin chains → the excess unpaired chains (especially α-chains in β-thalassemia) precipitate within red cell precursors, causing membrane damage and ineffective erythropoiesis + shortened RBC survival
In β-thalassemia major, massive ineffective erythropoiesis drives erythroferrone release, which suppresses hepcidin and causes secondary iron overload - the opposite of IDA. - Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 602-607
2. Etiology / Genetics
| Feature | IDA | Thalassemia |
|---|---|---|
| Cause | Iron depletion (nutritional, blood loss, malabsorption) | Inherited gene mutations |
| Inheritance | Acquired (not genetic) | Autosomal codominant |
| Gene | N/A | α-globin (chr 16), β-globin (chr 11) |
| Mutation type | N/A | β: mostly point mutations (splice, promoter, chain-terminator); α: mostly gene deletions |
| High-risk populations | Toddlers, adolescent girls, women of childbearing age, pregnancy | Mediterranean, Middle East, Africa, Southeast Asia, Indian subcontinent |
3. CBC and Laboratory Findings
This is the critical differentiating zone.
| Parameter | IDA | Thalassemia Trait | Key Point |
|---|---|---|---|
| RBC count | Low (~3.9 × 10⁶/µL) | High/Normal (>5.5 × 10⁶/µL men) | RBC elevated in thalassemia despite low MCV |
| Hemoglobin | Significantly low (~8 g/dL) | Mildly low (~12 g/dL) | IDA more severe |
| MCV | Low (~74 fL) | Lower (~69 fL for α-thal; 55-65 fL for β-thal) | Both microcytic |
| RDW | Markedly elevated (~19%) | Normal to mildly elevated (~14%) | Key differentiator |
| Platelets | Often elevated (reactive thrombocytosis) | Typically normal | Useful clue |
- Quick Compendium of Clinical Pathology, p. 218 (Table t4.4)
Iron Studies
| Test | IDA | Thalassemia |
|---|---|---|
| Serum iron | Low | Normal |
| TIBC | High | Normal |
| Transferrin saturation | Low (<15%) | Normal |
| Serum ferritin | Low (<12 µg/L) | Normal or elevated |
| Hepcidin | Low (suppressed) | Low (paradoxically, due to ineffective erythropoiesis) - but iron stores high |
The single most important differentiator in the clinic: normal serum iron + hypochromic microcytic anemia = thalassemia until proven otherwise. - Rosen's Emergency Medicine
4. Peripheral Blood Smear
IDA: Hypochromic microcytic cells with a wide zone of central pallor (pallor zone >1/3 diameter), marked poikilocytosis (elliptocytes, pencil cells), and anisocytosis (reflected in the high RDW).
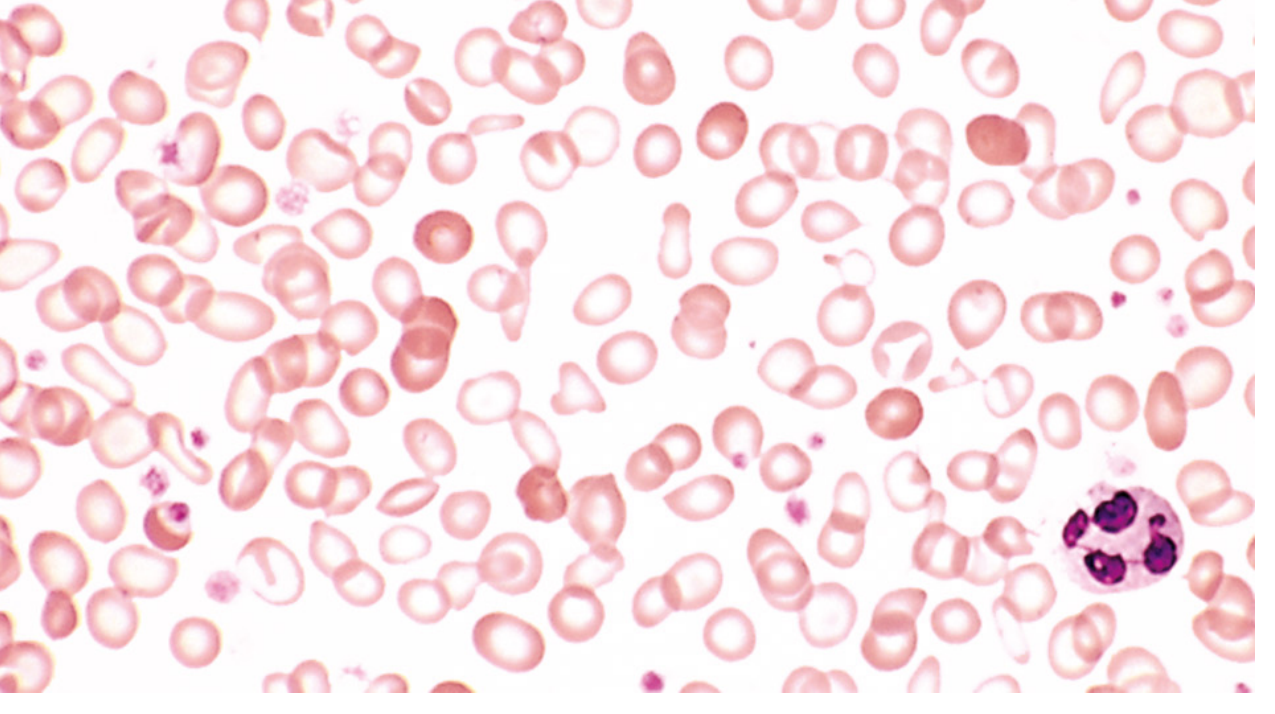
Thalassemia major: Target cells (codocytes), nucleated RBCs, basophilic stippling, Heinz bodies (from precipitated globin chains), marked poikilocytosis. In thalassemia trait, smear shows uniform microcytosis with minimal anisocytosis - Harrison's notes that in thalassemia the small cells are of uniform size with a normal-small RDW, whereas in IDA size variability is high.
5. Index-Based Discrimination
The Mentzer Index (MCV ÷ RBC count) is a quick bedside tool:
- < 13 → favors thalassemia
- > 15 → favors IDA
- 13-15 → indeterminate
Example: MCV 70 fL ÷ RBC 6.0 = 11.7 → thalassemia. MCV 70 fL ÷ RBC 3.8 = 18.4 → IDA.
- Quick Compendium of Clinical Pathology, p. 218
Other discriminant indices exist (England-Fraser, Shine-Lal, Green-King) but Mentzer remains the most widely used.
6. Clinical Features
| Feature | IDA | Thalassemia Major |
|---|---|---|
| Onset | Any age; insidious | 6-9 months of life (as HbF → HbA switch) |
| Severity | Mild-moderate typically | Severe: Hb 3-6 g/dL untransfused |
| Splenomegaly | Absent/mild | Prominent (extramedullary hematopoiesis) |
| Hepatomegaly | Absent | Present |
| Skeletal changes | Absent | "Crew-cut" skull x-ray, "chipmunk facies" from marrow expansion |
| Iron overload | No - iron depleted | Yes - secondary hemochromatosis (heart/liver damage) |
| Growth retardation | Absent | Present in untreated major |
| Pica / Pagophagia | Present (craving for ice, clay) | Absent |
| Koilonychia (spoon nails) | Present in severe/chronic IDA | Absent |
| Atrophic glossitis | Present | Absent |
| Response to oral iron | Reticulocytosis in 5-7 days | No response |
7. Hemoglobin Electrophoresis
This is the confirmatory test for thalassemia:
| Finding | Interpretation |
|---|---|
| Increased HbA₂ (>3.5%) | β-thalassemia trait - most reliable marker |
| Increased HbF | β-thalassemia major/intermedia |
| Normal HbA₂ + normal HbF + microcytosis | Suspect α-thalassemia (requires DNA analysis or Hb H detection) |
| Normal electrophoresis | Consistent with IDA (no abnormal hemoglobin) |
Note: IDA can spuriously lower HbA₂ levels, potentially masking β-thalassemia trait. Always correct iron deficiency before interpreting electrophoresis.
8. Causes Summary
IDA causes (by category):
- Dietary deficiency (low-resource settings, vegetarians, toddlers)
- Chronic blood loss (GI - colorectal cancer, peptic ulcer; menorrhagia)
- Malabsorption (celiac disease, post-gastrectomy)
- Increased demand (pregnancy, growth spurts)
- Hookworm infestation (tropical areas)
Thalassemia - purely genetic, no environmental cause. Heterozygous carriers (trait) are typically asymptomatic or mildly anemic. Homozygous β-thalassemia major requires lifelong transfusions.
9. Quick Summary Table
| Feature | IDA | Thalassemia |
|---|---|---|
| Type | Acquired nutritional | Inherited genetic |
| RBC count | Low | Normal/High |
| RDW | High | Normal/mildly high |
| Serum ferritin | Low | Normal/high |
| Serum iron | Low | Normal |
| TIBC | High | Normal |
| Mentzer index | >15 | <13 |
| Hb electrophoresis | Normal | Abnormal (↑ HbA₂/HbF in β-thal) |
| Response to iron | Yes | No |
| Peripheral smear | Anisocytosis, pencil cells | Uniform microcytes, targets, NRBCs |
| Spleen | Normal | Enlarged |
| Iron overload | Never | Common in major/severe forms |
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease (2024), pp. 602-615 | Quick Compendium of Clinical Pathology, 5th ed., p. 218 | Harrison's Principles of Internal Medicine 22E | Rosen's Emergency Medicine
This is a shared conversation. Sign in to Orris to start your own chat.