Functional anatomy of respiratory system
Reading File
Finding Sources
Finding Sources
Searching the Web
functional anatomy of respiratory system overview 2024
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Loading Image
Reading File
Reading File
I now have comprehensive content from Murray & Nadel's Textbook of Respiratory Medicine. Let me compile the full, detailed answer.
Functional Anatomy of the Respiratory System
(Source: Murray & Nadel's Textbook of Respiratory Medicine, 2-Volume Set)
Overview
The respiratory system's primary function is gas exchange - delivering O2 to the circulation and removing CO2 as a metabolic waste product. Its functional anatomy is organized into three integrated systems: (1) the airway tree, (2) the ventilatory pump (muscles, chest wall, neural control), and (3) the pulmonary vasculature.
1. The Airway Tree
Conducting Zone (Generations 0-16)
The airways form a branching tree that begins at the trachea (generation 0) and divides dichotomously at each generation, becoming progressively narrower, shorter, and more numerous. The first 16 generations - from trachea through bronchi and bronchioles to terminal bronchioles - constitute the conducting zone.
These airways contain no alveoli and take no part in gas exchange. Their function is to channel inspired gas to the gas-exchanging regions. Because they do not participate in gas exchange, they form the anatomic dead space (~150 mL in a normal adult).
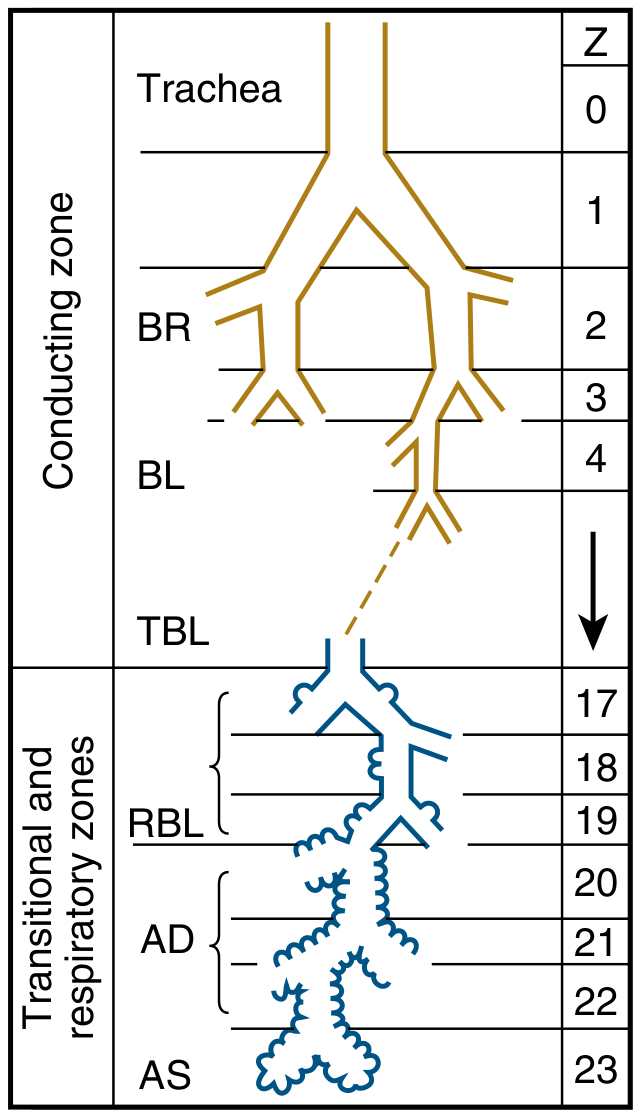
Key structural components:
- Trachea (Z=0): ~11 cm long, ~1.8 cm diameter, supported by C-shaped cartilage rings
- Main bronchi (Z=1): Right bronchus is more vertical, shorter, and wider - explains why aspirated foreign bodies tend to enter the right lung
- Lobar and segmental bronchi (Z=2-4): Supply individual lobes and bronchopulmonary segments
- Bronchioles (Z=5-16): Lack cartilage; walls maintained by elastic tissue and smooth muscle. Terminal bronchioles are the last purely conducting airways
Transitional Zone (Generations 17-19) - Respiratory Bronchioles
Respiratory bronchioles begin to have alveoli budding from their walls, giving them a dual role - partly conducting, partly gas-exchanging. The degree of alveolation steadily increases with each generation.
Respiratory Zone (Generations 20-23) - The Acinus
Each terminal bronchiole leads into a respiratory unit (acinus) - the functional unit of the lung. The acinus consists of:
- Respiratory bronchioles (Z=17-19): intermittent alveoli in walls
- Alveolar ducts (Z=20-22): walls are completely lined with alveoli
- Alveolar sacs (Z=23): blind-ending clusters of alveoli
The respiratory zone makes up the vast majority of the lung by volume (~2-3 L). Although the distance from terminal bronchiole to the most distal alveolus is only ~5 mm, this zone contains approximately 300 million alveoli with a total surface area of ~70 m².
Gas Transport: Convection to Diffusion
A key functional feature is the shift in gas transport mode at the level of the terminal bronchioles. Proximally, gas moves by bulk (convective) flow. Because total cross-sectional area increases enormously at generation 16-17 (resembling an inverted trumpet), forward gas velocity drops dramatically, and molecular diffusion takes over as the dominant mode of transport in the respiratory zone. This is highly efficient given the extremely short diffusion distances involved.
2. The Alveolar-Capillary Unit
The alveolus is the final functional unit for gas exchange. The blood-gas barrier consists of:
- Type I pneumocytes (thin, flat; 95% of alveolar surface) - primary gas exchange cells
- Basement membrane
- Pulmonary capillary endothelium
Type II pneumocytes (granular, cuboidal; 5% of surface) produce surfactant, which reduces surface tension, prevents alveolar collapse at end-expiration, and maintains alveolar stability across different lung volumes.
3. The Ventilatory Pump
The ventilatory pump consists of the neural controllers, conducting pathways, chest wall, and respiratory muscles.
Neural Control Architecture
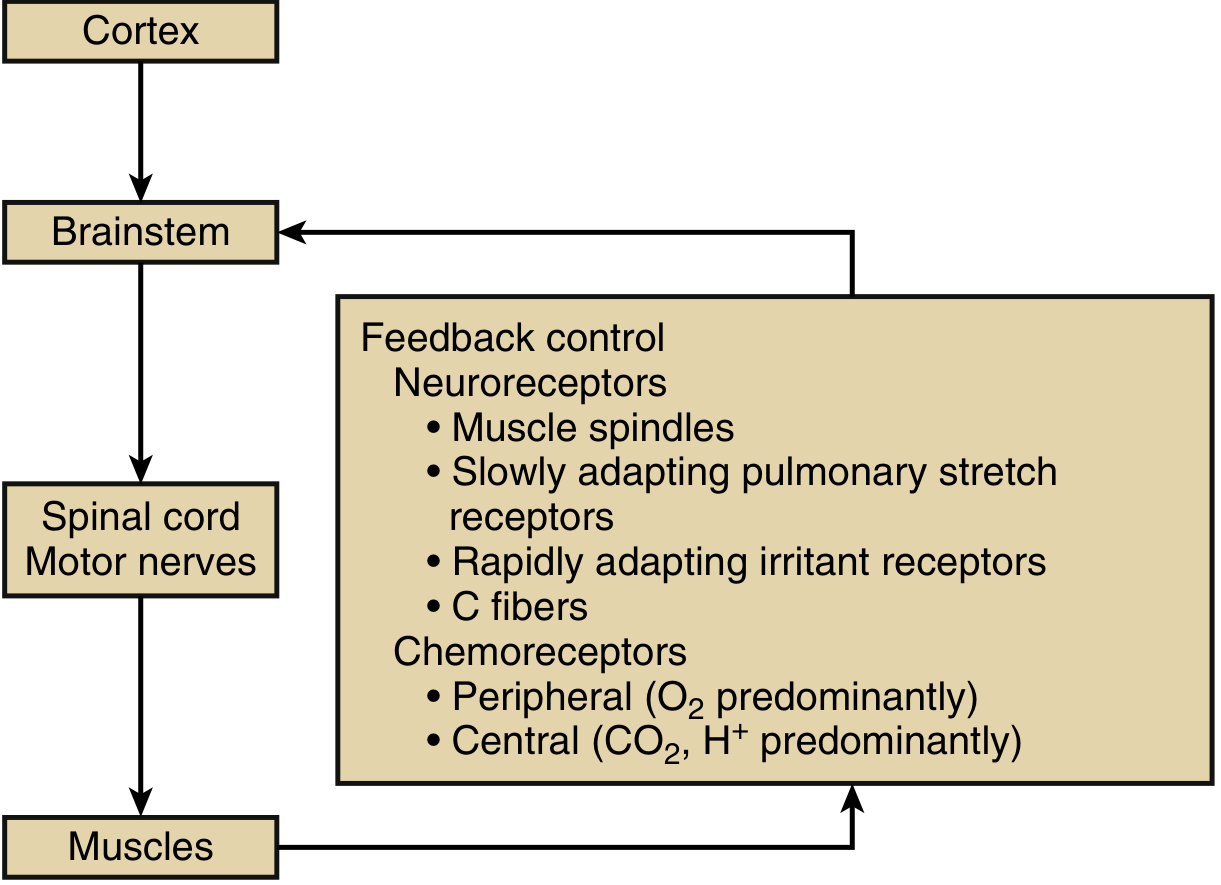
Voluntary control: The cerebral cortex (parietal cortex) initiates voluntary inspiration/expiration. Signals travel via the corticospinal tract to anterior horn motor neurons.
Automatic control: Brainstem respiratory centers (pons + medulla) generate the automatic breathing rhythm. Three centers are involved:
- Pneumotaxic center (pons): fine-tunes respiratory rate and limits tidal volume
- Apneustic center (pons): promotes sustained inspiration
- Pre-Bötzinger complex (medulla): the primary rhythm generator
Automatic signals travel via reticulospinal tracts - separate from the voluntary corticospinal pathways. This anatomical separation explains conditions like Ondine's curse (automatic breathing fails but voluntary breathing is preserved).
Spinal pathways: Motor neurons in the anterior horn cells receive both corticospinal and reticulospinal input and project to respiratory muscles via motor nerves.
Respiratory Muscles
Diaphragm - the principal muscle of inspiration, responsible for ~70% of inhaled tidal volume. Contraction produces a piston-like downward displacement, increasing thoracic volume and pushing lower ribs up and outward via the zone of apposition. Innervated by the phrenic nerve (C3-C5).
External intercostals - elevate ribs during inspiration, increasing antero-posterior and transverse thoracic diameter.
Internal intercostals - assist forced expiration by depressing ribs (opposite action to externals).
Accessory muscles (used during increased ventilatory demand):
- Sternocleidomastoid, scalenes - elevate upper rib cage on inspiration
- Abdominal muscles - the primary expiratory muscles; active in forced expiration, coughing, and sneezing
Normal quiet expiration is passive, driven by elastic recoil of the lungs and chest wall.
Feedback Receptors
The system maintains precise homeostasis via multiple receptor types:
| Receptor | Location | Stimulus | Function |
|---|---|---|---|
| Slowly adapting stretch receptors | Airway smooth muscle | Lung inflation | Hering-Breuer reflex - stops inspiration as lung inflates |
| Rapidly adapting irritant receptors | Airway epithelium | Dust, chemicals, volume change | Cough, sneeze, bronchoconstriction |
| C fibers (J receptors) | Lung parenchyma/pulmonary vessels | Chemical stimuli, edema | Rapid shallow breathing, dyspnea |
| Muscle spindles | Respiratory muscles | Muscle stretch | Monitor effort vs. displacement |
| Peripheral chemoreceptors (carotid/aortic bodies) | Carotid bifurcation, aortic arch | Low PaO2 (<75 mmHg), rising PaCO2, falling pH | Primary O2 sensors; carotid bodies dominant in adults |
| Central chemoreceptors | Ventrolateral medulla, NTS, raphe | Rising PaCO2/falling CSF pH | Primary CO2/acid-base sensors; drive most of resting ventilatory tone |
4. Lung Volumes and Functional Compartments
| Volume/Capacity | Definition | Normal Value |
|---|---|---|
| Tidal Volume (VT) | Air per breath at rest | ~500 mL |
| Inspiratory Reserve Volume (IRV) | Extra air beyond normal inspiration | ~3000 mL |
| Expiratory Reserve Volume (ERV) | Extra air beyond normal expiration | ~1100 mL |
| Residual Volume (RV) | Air remaining after max expiration | ~1200 mL |
| Functional Residual Capacity (FRC) | ERV + RV; lung volume at end-expiration | ~2300 mL |
| Total Lung Capacity (TLC) | All lung air at max inspiration | ~5800 mL |
| Vital Capacity (VC) | TLC - RV | ~4600 mL |
| Anatomic Dead Space | Conducting airways volume | ~150 mL |
5. Distribution of Ventilation
Ventilation is not uniform throughout the lung. In the upright position, ventilation per unit volume is greatest at the base and decreases toward the apex. This results from gravity-dependent pleural pressure gradients:
- Intrapleural pressure is less negative at the lung base (due to lung weight)
- Basal alveoli are more compressed at FRC but have greater compliance slope - so they receive more ventilation per breath
- Apical alveoli are more expanded at FRC and sit on a flatter part of the pressure-volume curve
This regional inequality of ventilation, when mismatched with perfusion (V/Q mismatch), is the most common cause of hypoxemia in clinical disease.
Summary of Functional Zones
UPPER RESPIRATORY TRACT LOWER RESPIRATORY TRACT
Nose → Pharynx → Larynx | Trachea → Bronchi → Bronchioles
| ↓
CONDUCTING ZONE (Z 0-16): Dead space, gas conditioning
↓
TRANSITIONAL ZONE (Z 17-19): Respiratory bronchioles
↓
RESPIRATORY ZONE (Z 20-23): Alveolar ducts & sacs
← Gas exchange occurs here →
The elegance of the respiratory system lies in its division of labor: the upper tract warms, humidifies, and filters inspired air; the conducting zone channels gas efficiently with near-zero resistance cost; and the respiratory zone provides an enormous surface area (70 m²) at a vanishingly thin barrier (<0.5 μm) to enable the 300 mL/min of O2 transfer needed at rest, scalable to >3 L/min during heavy exercise.
References: Murray & Nadel's Textbook of Respiratory Medicine, 2-Volume Set - Chapters 10 (Gas Exchange) and 130 (Functional Anatomy / Neuromuscular Respiratory System)
Search for recent research on surfactant dysfunction in ARDS
Reading File
Searching PubMed
surfactant dysfunction ARDS
Searching PubMed
surfactant therapy acute respiratory distress syndrome
Finding Sources
Reading Article
PMID: 40961332
Reading Article
PMID: 36551272
Reading Article
PMID: 41892849
Reading File
Reading File
Searching PubMed
surfactant replacement therapy adult ARDS outcome
Reading File
Reading File
I now have comprehensive textbook content and recent PubMed literature. Let me compile the full answer.
Recent Research on Surfactant Dysfunction in ARDS
Background: Surfactant Composition and Normal Function
Surfactant is a lipoprotein complex secreted by type II alveolar epithelial (AT2) cells. Its critical lipid component is dipalmitoylphosphatidylcholine (DPPC) (~50-60% of surfactant lipid), which forms a tightly packed monolayer at the air-liquid interface. Four associated proteins fine-tune its function:
| Protein | Type | Function |
|---|---|---|
| SP-A | Hydrophilic | Immune regulation, opsonization, tubular myelin formation |
| SP-B | Hydrophobic | Lipid film adsorption and spreading; essential for life |
| SP-C | Hydrophobic | Lipid insertion into surface monolayer |
| SP-D | Hydrophilic | Innate immunity, pathogen agglutination, inflammation regulation |
Together, these components reduce alveolar surface tension to near 0 mN/m at end-expiration, preventing alveolar collapse and stabilizing alveoli across a range of sizes (Law of Laplace). (Murray & Nadel's Respiratory Medicine, Ch. 3)
How ARDS Disrupts Surfactant
ARDS triggers surfactant dysfunction through several overlapping mechanisms, extensively described in both textbooks and recent literature:
1. Reduced Surfactant Production
Inflammatory mediators in ARDS cause AT2 cell death and dysfunction, directly reducing surfactant synthesis and secretion. AT1 cell loss exposes the basement membrane, while surviving AT2 cells are partially resistant to injury but functionally impaired. The impairment of surfactant production is listed alongside disrupted intercellular junctions and impaired Na+/K+ pump function as core mechanisms of alveolar cell injury in ARDS. (Murray & Nadel, Ch. ARDS Pathogenesis)
2. Surfactant Inactivation in the Alveolar Space
Increased alveolar-capillary permeability in ARDS floods the air spaces with protein-rich edema fluid. Plasma proteins - particularly albumin, fibrinogen, and C-reactive protein - competitively inhibit surfactant at the air-liquid interface by displacing phospholipids from the surface monolayer. The result: surface tension rises, alveoli collapse, shunt increases, and hypoxemia worsens. (Murray & Nadel, Ch. 3 & 11)
3. Altered Surfactant Composition
The ratio of surfactant components shifts unfavorably in ARDS:
- SP-B and SP-C levels decrease significantly
- Oxidized phospholipid species accumulate (dysfunctional)
- The phosphatidylcholine/phosphatidylglycerol ratio is altered
- Large aggregate (functional) surfactant is converted to small aggregate (non-functional) forms by inflammatory phospholipases
4. Functional Consequences
"Surfactant dysfunction increases surface tension at the air-liquid interface of the alveoli, leading to collapse, intrapulmonary shunting, and hypoxemia." (Murray & Nadel, Recruitment Maneuvers in ARDS)
Recent Literature (2022-2026)
[Review · 2026] Hyperoxia and Surfactant Dysfunction in Critical Illness
Watson A, Roe T, Terrington I et al. Am J Respir Cell Mol Biol [PMID: 40961332]
This 2026 review specifically addresses hyperoxia-induced surfactant dysregulation - an underappreciated driver in critically ill patients requiring high FiO2. Key findings and concepts:
- Hyperoxia directly disrupts surfactant lipids and proteins (particularly SP-A and SP-D), impairs surfactant synthesis and metabolism, and promotes atelectasis
- Redox imbalances and phospholipase A2 activation degrade surfactant phospholipids under oxidative stress
- Altered macrophage clearance of surfactant debris is proposed as a novel mechanism
- Hyperoxia-induced injury interacts synergistically with mechanical ventilation injury (VILI) and infection-driven ARDS
- The review calls for novel surfactant preparations resistant to functional inhibition and better human models to distinguish oxygen dose/duration effects
Key clinical implication: even when systemic SpO2 is maintained, alveolar hyperoxia from supplemental O2 may worsen surfactant function - supporting restrictive oxygen strategies in ARDS management.
[Review · 2026] Clinical, Molecular, and Therapeutic Approaches to ARDS
Gonzalez-Plascencia M et al. Med Sci (Basel) [PMID: 41892849]
This 2026 comprehensive review frames surfactant dysfunction within the broader biology of ARDS:
- Describes ARDS as characterized by "epithelial and endothelial injury, dysregulated inflammation, surfactant dysfunction, and impaired alveolar-capillary barrier integrity"
- Emphasizes biological heterogeneity across ARDS phenotypes - surfactant dysfunction is more prominent in the hyperinflammatory phenotype ("hot ARDS")
- Advocates for biomarker-based ARDS phenotyping to identify patients most likely to benefit from surfactant-directed therapies
- Notes precision-based approaches as the way forward, away from syndromic management
[Review · 2022] Surfactant Dysfunction in COVID-19/ARDS
Krygier A et al. Biomolecules [PMID: 36551272]
This review systematically details three key lung pathologies in severe COVID-19 ARDS:
- SARS-CoV-2 directly infects AT2 cells (which express ACE2), causing direct surfactant deficiency - distinct from secondary inactivation seen in classic ARDS
- Concurrent activation of the coagulation cascade and fibroblast proliferation creates fibrin plugs that further inactivate surfactant in the alveolar space
- Pulmonary fibrosis with myofibroblast proliferation and collagen deposition further impairs alveolar mechanics
- Offers insight into why COVID-ARDS had a distinct phenotype from classical ARDS, with deeper surfactant deficiency and worse response to conventional ventilatory strategies
Surfactant Therapy Trials: Why Adult ARDS Remains Challenging
Despite the clear pathophysiology, exogenous surfactant therapy has not demonstrated mortality benefit in adults with ARDS across multiple trials. The principal obstacles identified in recent research:
| Challenge | Detail |
|---|---|
| Delivery | Intratracheal instillation cannot distribute evenly in injured, flooded lungs |
| Inactivation | Exogenous surfactant is rapidly inhibited by the same plasma proteins causing endogenous dysfunction |
| Dose | The dose required to overcome protein inhibition in adults far exceeds what's feasible |
| Heterogeneity | ARDS is a syndrome with multiple phenotypes; not all have equivalent surfactant depletion |
| Composition | Current animal-derived preparations lack full complement of SP-A/SP-D |
A retrospective case-control pilot study of surfactant therapy for COVID-ARDS (Piva S et al., 2021, PMID: 33461535) suggested potential benefit in selected patients, but sample sizes were too small for conclusions.
Emerging Therapeutic Directions
Based on the 2026 literature, the most promising directions are:
- Novel surfactant formulations resistant to protein inactivation - synthetic or semi-synthetic preparations that maintain function in protein-rich environments
- SP-B/SP-C mimetics - peptide analogues of the hydrophobic surfactant proteins with improved inactivation resistance
- Biomarker-guided patient selection - using SP-D, SP-B levels, or phenotypic clustering (hyper- vs. hypo-inflammatory) to identify ARDS patients with the greatest surfactant deficiency who may respond to replacement
- Oxygen-sparing strategies - targeting SpO2 94-96% rather than >98% to reduce alveolar hyperoxia-mediated surfactant degradation (supported by the 2026 Watson et al. review)
- Phospholipase A2 inhibitors - targeting the enzyme responsible for surfactant phospholipid degradation under oxidative stress
Summary Table: Mechanisms of Surfactant Dysfunction in ARDS
| Mechanism | Trigger | Result |
|---|---|---|
| AT2 cell death/dysfunction | Inflammatory mediators, SARS-CoV-2 | Decreased surfactant synthesis |
| Plasma protein inhibition | Alveolar flooding (albumin, fibrinogen) | Displacement from air-liquid interface |
| Oxidized phospholipid accumulation | ROS, hyperoxia, neutrophil MPO | Dysfunctional surface film |
| Phospholipase A2 activation | Oxidative stress, cytokines | Phospholipid degradation |
| SP-B / SP-C depletion | Cell death, inhibition | Poor film adsorption and spreading |
| Large-to-small aggregate conversion | Inflammatory milieu | Loss of functional surfactant pool |
Bottom line: Surfactant dysfunction in ARDS is multifactorial and well-established, but therapeutic exploitation remains elusive in adults. The 2026 literature highlights hyperoxia-driven surfactant degradation as a newly characterized and clinically actionable pathway, while precision phenotyping may help identify patients who could benefit from future surfactant-directed interventions.
This is a shared conversation. Sign in to Orris to start your own chat.