Metastasis and routes
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
I now have comprehensive, authoritative content from Robbins Pathology. Let me compile the full answer.
Metastasis and Routes of Spread
Metastasis is defined as the spread of a primary tumor to physically discontinuous sites - an event that unequivocally marks a tumor as malignant. Approximately 30% of solid tumors (excluding non-melanoma skin cancers) present with clinically evident metastases at diagnosis, and an additional 20% have occult metastases at that time. - Robbins & Kumar Basic Pathology, p. 221
Key Concept: Why Metastasis is Inefficient
Although many locally invasive tumor cells enter the bloodstream each day, very few produce actual metastases. For cancer cells to produce a secondary growth at a distant site, they must successfully complete a complex series of steps - evading immune defenses and adapting to a foreign microenvironment at each stage. This is why individual "metastasis genes" have not been found; the metastatic phenotype likely requires accumulation of multiple complementary genetic and epigenetic alterations. - Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 290
The Three Pathways (Routes) of Metastatic Spread
Dissemination of cancers occurs through three main pathways:
1. Seeding of Body Cavities and Surfaces
This occurs when malignant neoplasms invade a natural "open field" lacking physical barriers.
- Most commonly involves the peritoneal cavity
- Other surfaces affected: pleura, pericardium, meninges, and joints
- Classic example: Ovarian carcinoma - spreads widely over peritoneal surfaces yet may not invade the underlying tissues; often covers the peritoneal surfaces with a heavy cancerous coating
- Mucus-secreting appendiceal or ovarian carcinomas may fill the peritoneal cavity with gelatinous neoplastic mass = pseudomyxoma peritonei
- CNS tumors (e.g., medulloblastoma, ependymoma) may penetrate cerebral ventricles and be carried by cerebrospinal fluid to implant on meningeal surfaces
2. Lymphatic Spread
Transport through lymphatic vessels is the most common pathway for initial dissemination of carcinomas. Sarcomas can also use this route.
Key points:
- Tumors do not contain functional lymphatic vessels; lymphatics at the tumor margins are sufficient for spread
- The pattern of spread follows natural routes of lymphatic drainage
- Skip metastasis can occur - cancer cells bypass the immediately proximal nodes and are trapped in subsequent nodes (possibly due to variation in normal lymphatic drainage patterns)
- Sentinel lymph node = the first regional lymph node receiving lymph flow from a primary tumor; can be identified by injection of dyes or radiolabeled tracers near the tumor; biopsy determines extent of spread
Examples of lymphatic spread patterns:
| Primary Tumor | First Nodes Involved |
|---|---|
| Breast (upper outer quadrant) | Axillary nodes → infraclavicular/supraclavicular |
| Breast (medial lesions) | Internal mammary artery nodes |
| Lung (major airways) | Perihilar tracheobronchial and mediastinal nodes |
| Colon cancer | Follows major venous outflow from involved segment |
Note: Enlarged lymph nodes near a primary neoplasm do not always indicate cancerous involvement - necrotic tumor cells and tumor antigens can evoke reactive hyperplasia (lymphadenitis). Biopsy is necessary.
3. Hematogenous Spread
Most characteristic of sarcomas, though carcinomas also use this route (both lymphatic and vascular systems are interconnected).
Key points:
- Tumor cells preferentially penetrate thin-walled veins rather than thick-walled arteries
- Cells arrest in the first capillary bed encountered
- Portal blood flows to liver → liver is most common site
- Caval blood flows to lungs → lungs are most common site
- Renal cell carcinoma and hepatocellular carcinoma have a propensity to grow within veins in a snakelike fashion, extending up the IVC to the right heart
Organ-specific spread patterns (beyond anatomy):
| Tumor | Preferential Site | Mechanism |
|---|---|---|
| Prostate, thyroid | Vertebral column (spine) | Embolize via paravertebral plexus |
| Prostatic carcinoma | Bone | Organ-specific homing |
| Bronchogenic carcinoma | Adrenal glands, brain | Organ-specific homing |
| Neuroblastoma | Liver, bones | Organ-specific homing |
| Uveal melanoma | Liver | Organ-specific homing |
- Skeletal muscle, despite being rich in capillaries, is rarely a site of metastasis
The Metastatic Cascade (Hematogenous Route - Step by Step)
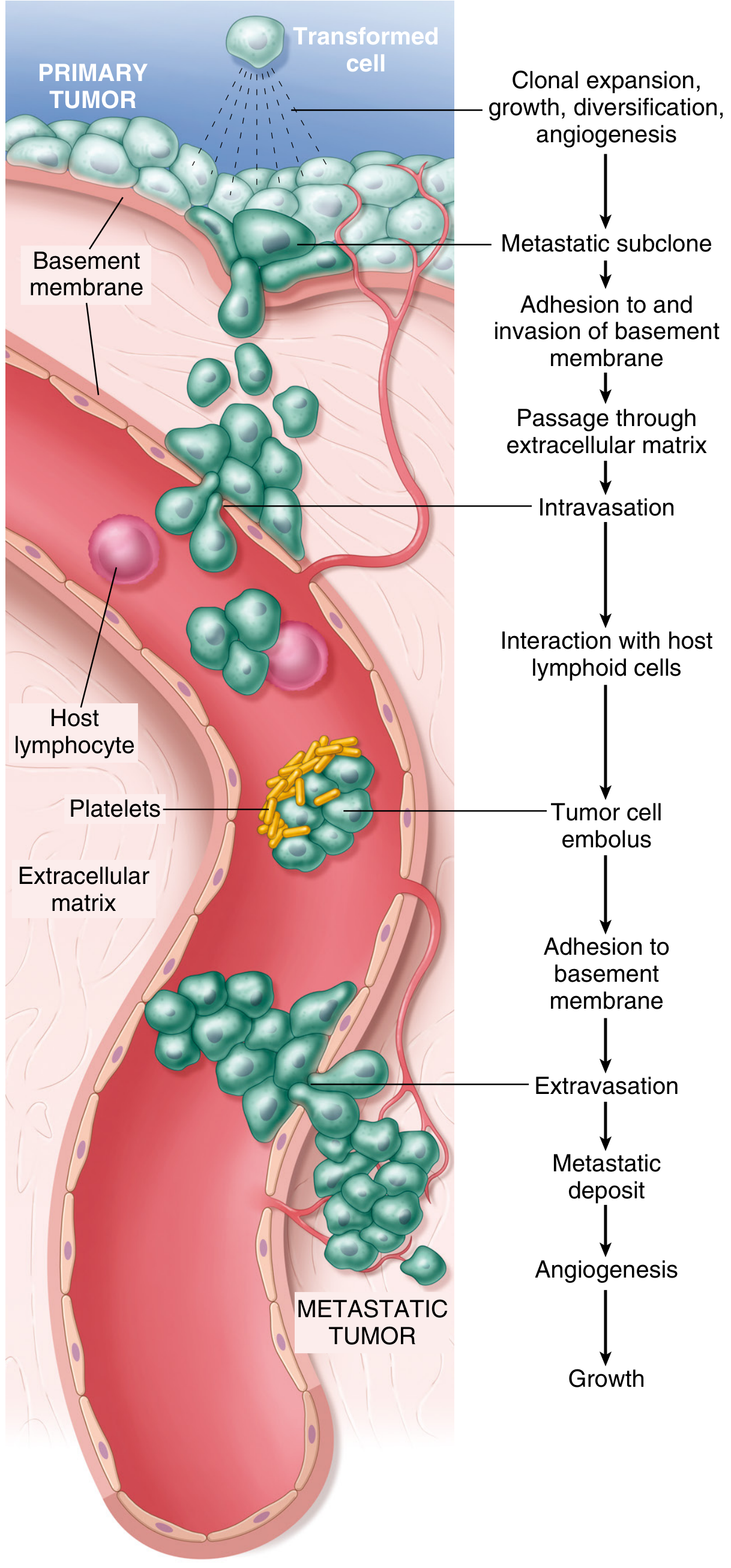
Fig. 7.36 - The metastatic cascade. Sequential steps in hematogenous spread.
The metastatic cascade is divided into two phases:
Phase 1: Invasion of the Extracellular Matrix (ECM)
Step 1 - Loosening of tumor cell-tumor cell interactions:
- E-cadherin (transmembrane glycoproteins) function is lost due to mutations
- Epithelial-Mesenchymal Transition (EMT) - silences E-cadherin expression
- Controlled by transcription factors SNAIL and TWIST
- Downregulates epithelial markers (E-cadherin)
- Upregulates mesenchymal markers (vimentin, smooth muscle actin)
- Creates a promigratory phenotype
Step 2 - Degradation of ECM:
- Tumor cells secrete proteolytic enzymes OR induce stromal cells (fibroblasts, inflammatory cells) to produce them
- Key enzymes: Matrix metalloproteinases (MMPs), cathepsin D, urokinase plasminogen activator
- MMP9 (gelatinase): cleaves type IV collagen in basement membranes; also releases VEGF from ECM-sequestered pools
- Benign tumors have little MMP9 activity vs. malignant counterparts which overexpress it
- Metalloproteinase inhibitor concentrations are also reduced in many cancers
Step 3 - Attachment to remodeled ECM components:
- Tumor cells bind to newly exposed ECM binding sites created by protease cleavage
Step 4 - Migration and invasion:
- Tumor cells move through degraded basement membrane and interstitial connective tissue
- Guided by chemotactic factors produced by inflammatory and stromal cells
Phase 2: Vascular Dissemination, Tissue Homing, and Colonization
- Intravasation - tumor cells penetrate blood vessel/lymphatic walls
- Circulation as tumor cell emboli - often coated with platelets (protective)
- Interaction with host lymphoid cells - surviving immune surveillance
- Arrest in distant capillary bed - adhesion to basement membrane
- Extravasation - exit from blood vessel
- Formation of metastatic deposit - colonization of new tissue
- Angiogenesis and growth at metastatic site
Liver Studded with Metastatic Cancer (Gross Pathology)
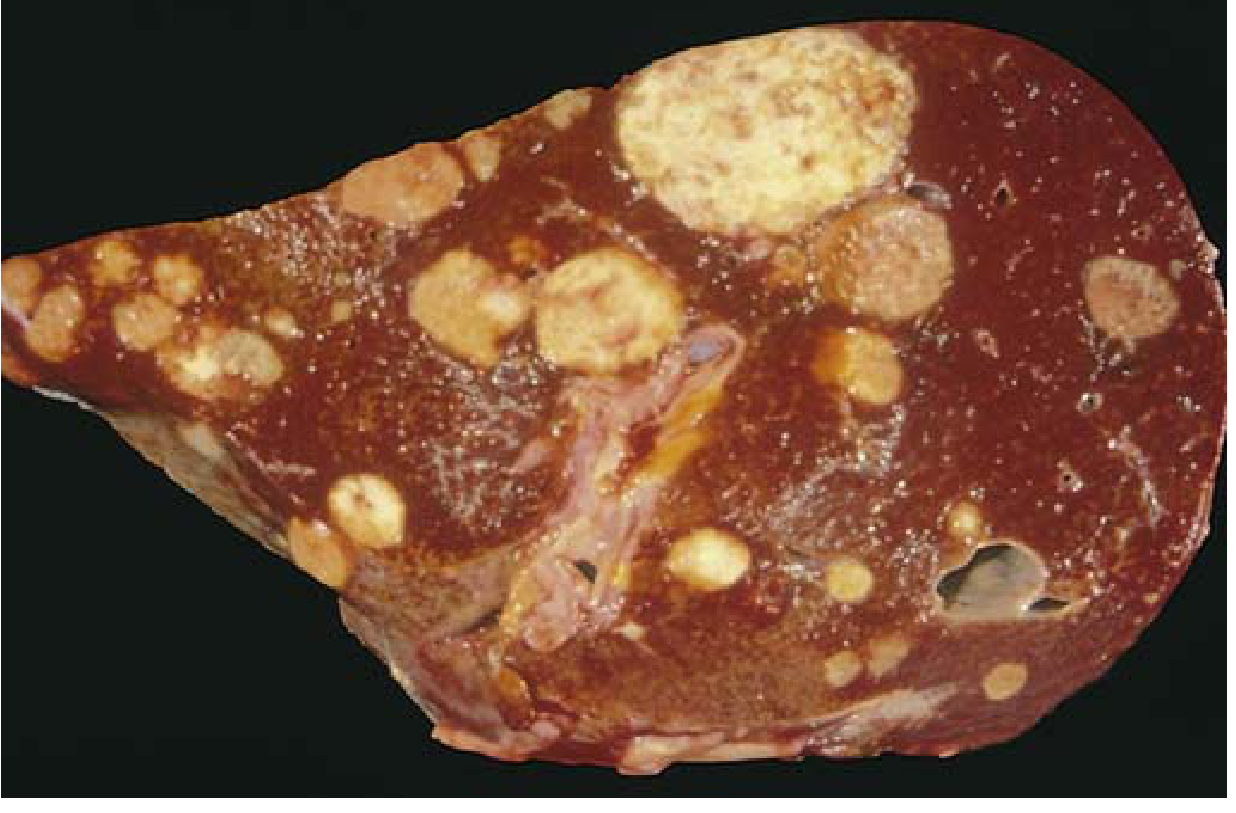
FIG. 6.9 - A liver studded with metastatic cancer - classic appearance of hematogenous spread via portal system.
Tumors That Rarely Metastasize
Despite being locally invasive:
- Gliomas (malignant CNS tumors) - rarely metastasize
- Basal cell carcinomas of skin - rarely metastasize
- This demonstrates that local invasiveness and metastatic potential are separable properties
Special Case: Leukemias and Lymphomas ("Liquid Tumors")
- Derived from blood-forming cells that normally circulate
- Assumed to be disseminated at diagnosis by definition
- Always considered malignant regardless of spread
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease (p. 259, 290-291); Robbins & Kumar Basic Pathology (p. 221-222)
Molecular basis of carcinogenesis
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Excellent - I have comprehensive content now. Let me compile the full answer.
Molecular Basis of Carcinogenesis
Carcinogenesis is fundamentally a genetic disease. All cancers arise from somatic mutations and epigenetic alterations that accumulate in genes governing normal cell behavior. The modern framework organizes these changes around the Hallmarks of Cancer - universal biologic properties that all cancers must acquire.
The Hallmarks of Cancer
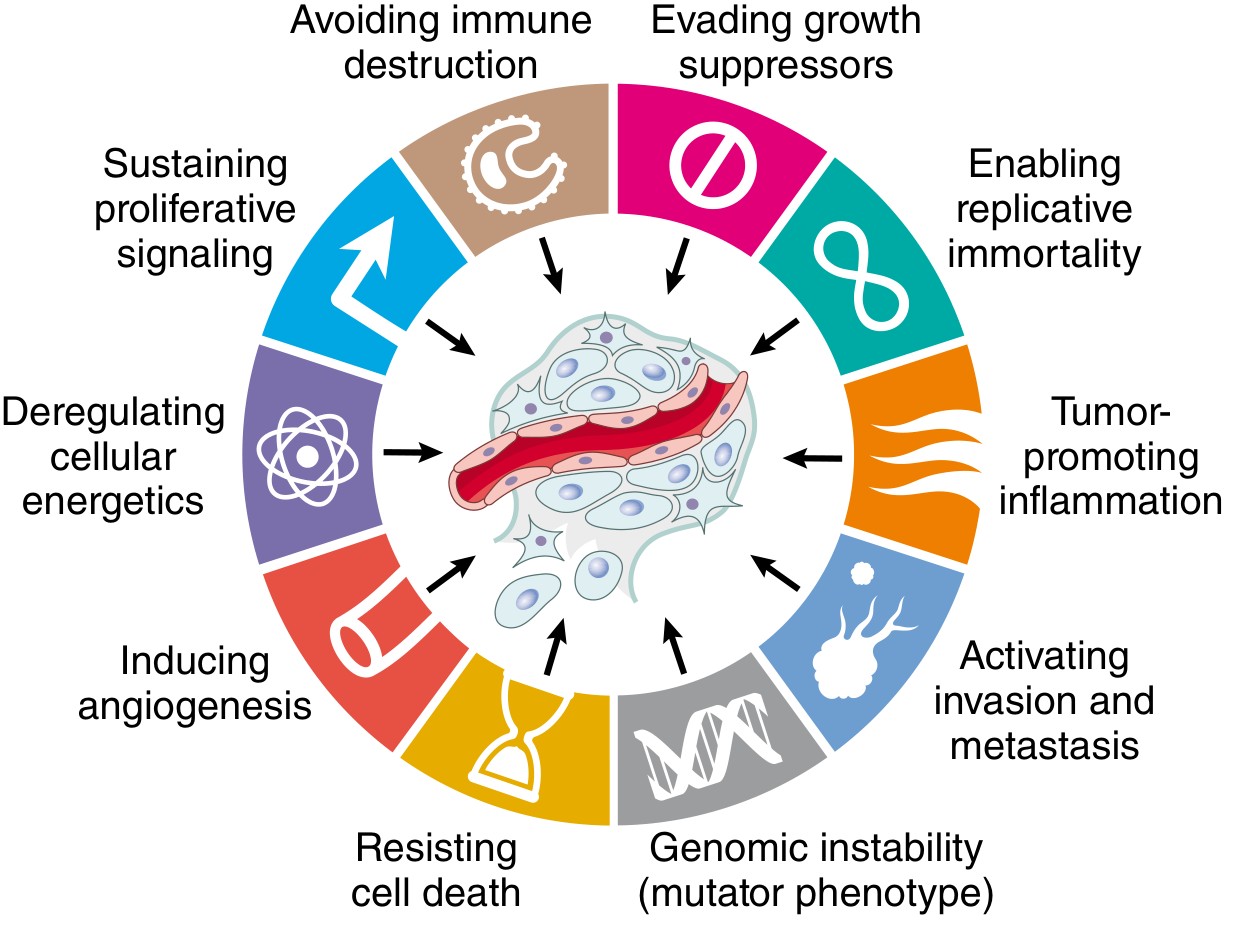
Fig. 7.20 - Hallmarks of Cancer (Hanahan & Weinberg). Eight fundamental changes in cell physiology.
All cancers display eight fundamental hallmarks, each driven by specific molecular alterations: - Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 251-268
| Hallmark | Molecular Basis |
|---|---|
| 1. Self-sufficiency in growth signals | Oncogene activation |
| 2. Insensitivity to growth-inhibitory signals | Tumor suppressor gene inactivation |
| 3. Altered cellular metabolism (Warburg effect) | Switch to aerobic glycolysis |
| 4. Evasion of apoptosis | Anti-apoptotic gene alterations |
| 5. Limitless replicative potential (immortality) | Telomerase activation |
| 6. Sustained angiogenesis | VEGF overexpression |
| 7. Ability to invade and metastasize | ECM degradation, EMT |
| 8. Evasion of host immune response | Immune checkpoint exploitation |
An enabling feature underlying all hallmarks: genomic instability (mutator phenotype) - accelerates accumulation of driver mutations.
I. Types of Cancer Genes
Cancer genes fall into two fundamental categories based on the behavior of their mutations:
A. Proto-Oncogenes → Oncogenes (Gain-of-function)
- Proto-oncogenes: Normal cellular genes whose products promote cell proliferation
- Oncogenes: Mutated or overexpressed versions of proto-oncogenes that function autonomously - having lost dependence on normal growth-promoting signals
- Oncoproteins: Proteins encoded by oncogenes that drive increased cancer cell proliferation
Oncogenic mutations are dominant - a single mutated allele is sufficient to drive abnormal behavior.
How Proto-Oncogenes Become Oncogenes:
1. Point mutations - e.g., RAS point mutations (most common, present in many cancers)
2. Gene amplification - e.g., HER2/ERBB2 amplification in breast cancer; MYC amplification in neuroblastoma; N-MYC in small cell lung cancer
3. Chromosomal translocations - e.g.:
- BCR::ABL fusion (t(9;22), Philadelphia chromosome) → Chronic Myeloid Leukemia (CML)
- MYC overexpression via t(8;14) → Burkitt lymphoma
- BCL2 overexpression via t(14;18) → Follicular lymphoma (anti-apoptotic)
4. Overexpression - autocrine growth factor loops (gliomas overexpressing PDGF + PDGFR)
Key Oncoproteins by Category:
Growth Factors:
- PDGF-β → astrocytoma
- TGF-α → many carcinomas (autocrine loop)
Growth Factor Receptors (Receptor Tyrosine Kinases):
- EGFR - point mutations → lung adenocarcinoma (targetable with gefitinib/erlotinib)
- HER2/ERBB2 - gene amplification → breast cancer (targetable with trastuzumab)
- RET - point mutations → multiple endocrine neoplasia (MEN) type 2
- Oncogenic RTK mutations cause constitutive, growth factor-independent tyrosine kinase activity
Non-receptor Tyrosine Kinases:
- ABL → BCR::ABL fusion → CML (targetable with imatinib)
- JAK2 → point mutations → myeloproliferative neoplasms
Signal Transducers (G proteins):
- RAS (KRAS, NRAS, HRAS) → mutated in ~30% of all cancers
- Normally: GDP-bound (inactive) ↔ GTP-bound (active)
- Oncogenic mutation: blocks GTPase activity → constitutively GTP-bound = constitutively active
- Activates downstream MAPK and PI3K/AKT pathways
- BRAF (downstream of RAS) → point mutations → melanoma, colorectal cancer
Transcription Factors:
- MYC - master transcription factor; regulates genes for rapid cell growth
- Overexpressed via chromosomal translocations (Burkitt), gene amplification (neuroblastoma)
- Drives expression of cyclins → cell cycle progression
Cell Cycle Components:
- CDK4 overexpression/mutation → accelerates G1→S transition (many cancers)
- Cyclin D1 amplification → breast, esophageal cancers
B. Tumor Suppressor Genes (Loss-of-function)
Tumor suppressor gene products act as negative regulators of cell proliferation. Loss of their function leads to excessive growth. Mutations are recessive at the cellular level - both alleles must be lost.
Knudson's Two-Hit Hypothesis (RB gene prototype):
- Familial retinoblastoma: inherit one defective RB allele (1st hit) + acquire somatic mutation in normal allele (2nd hit) → tumor
- Sporadic retinoblastoma: two independent somatic mutations required → rarer, usually unilateral
The RB Gene (Retinoblastoma protein)
RB is the gatekeeper of the cell cycle G1→S checkpoint:
- Hypophosphorylated RB (active form): binds and inhibits the E2F transcription factor → blocks cell cycle progression in G1
- Growth factor signaling activates CDK4/6 + cyclin D → phosphorylates RB → releases E2F → transcription of S-phase genes → cell cycle entry
- Oncogene mutations (CDK4, cyclin D amplification) or loss of CDK inhibitors (p16/INK4a) all converge on RB inactivation
The TP53 Gene ("Guardian of the Genome")
p53 is the most frequently mutated gene in human cancers (>50% of tumors have TP53 mutations).
Normal function: p53 is the focal point of a large network that senses cellular stress (DNA damage, shortened telomeres, hypoxia, oncogenic signaling).
Regulation:
- In non-stressed cells: p53 is bound and ubiquitinated by MDM2 → proteasomal degradation (virtually undetectable)
- MDM2 gene is amplified in 33% of sarcomas → functional p53 deficiency without TP53 mutation
- HPV E6 protein binds p53 and promotes its degradation (mechanism in cervical carcinoma)
Activation pathways:
- DNA damage/hypoxia → ATM/ATR kinases phosphorylate p53 and MDM2 → disrupts p53-MDM2 binding → p53 accumulates
- Oncogenic stress (RAS activation) → p14/ARF (encoded by CDKN2A) → binds MDM2 → displaces p53
p53 Responses (once activated):
- Transient cell cycle arrest → DNA repair → resume division (if repair successful)
- Senescence (permanent cell cycle arrest)
- Apoptosis (programmed cell death) - via transcription of pro-apoptotic genes (BAX, PUMA)
Other Key Tumor Suppressor Genes:
| Gene | Protein/Function | Familial Syndrome | Sporadic Cancers |
|---|---|---|---|
| APC | Inhibits Wnt/β-catenin signaling | Familial adenomatous polyposis (FAP) | ~70% sporadic colon carcinomas |
| CDKN2A (p16/INK4a) | CDK inhibitor - augments RB function | Familial melanoma | Leukemias, melanomas, carcinomas |
| CDKN2A (p14/ARF) | Stabilizes p53 via MDM2 inhibition | Familial melanoma | Many cancers |
| PTEN | Lipid phosphatase - inhibits PI3K/AKT | Cowden syndrome | Breast, endometrial, prostate |
| VHL | Degrades HIF transcription factors | Von Hippel-Lindau syndrome | Sporadic renal cell carcinoma |
| E-cadherin (CDH1) | Cell adhesion, contact inhibition; sequesters β-catenin | Familial gastric carcinoma | Many sporadic carcinomas + increased invasiveness |
| NF1 | Inhibitor of RAS/MAPK signaling | Neurofibromatosis type 1 | Neuroblastoma, juvenile myeloid leukemia |
| BRCA1/BRCA2 | DNA repair | Hereditary breast/ovarian cancer | Sporadic breast, ovarian cancers |
| TGF-β receptors/SMADs | Potent inhibitor of proliferation | - | Stomach, endometrium, pancreas |
| STK11 | Serine/threonine kinase; metabolic regulator | Peutz-Jeghers syndrome | Lung, GI cancers |
II. Mechanisms of Oncogene Activation - Key Signaling Pathways
Normal physiologic growth factor signaling involves:
- Growth factor binds receptor
- Transient receptor activation → cytoplasmic signal transducers (RAS, PI3K)
- Signal transmission to nucleus
- Transcription factor activation
- Gene expression → cell cycle entry + proliferation
Oncogenic mutations short-circuit these controls at every level:
RAS-MAPK Pathway
- RAS mutation → constitutive MAPK activation → transcription of pro-proliferative genes (FOS, JUN, MYC)
PI3K-AKT-mTOR Pathway
- PI3K mutations/PTEN loss → constitutive AKT activation → promotes cell survival (inhibits apoptosis), protein synthesis, Warburg effect
Wnt-β-catenin Pathway
- APC loss or β-catenin mutations → β-catenin accumulates in nucleus → activates MYC, cyclin D1 → proliferation
III. Molecular Basis of Multistep Carcinogenesis
Cancer does not arise from a single mutation - it requires stepwise accumulation of multiple complementary mutations.
Evidence - Colorectal Carcinoma (classic model):
Normal epithelium
↓ APC mutation (early)
Small adenoma
↓ KRAS mutation
Larger adenoma
↓ SMAD4/TGF-β loss
↓ TP53 mutation (late)
Adenocarcinoma
- Certain mutations occur early (APC in colon cancer)
- Others occur late (TP53)
- Similar stepwise progression documented for cervical dysplasia, endometrial hyperplasia, epidermal dysplasia
Experimental evidence: Normal human epithelial cells can be fully transformed into malignant cells by combining: (1) RAS activation, (2) RB inactivation, (3) p53 inactivation, (4) PP2A inactivation, and (5) telomerase expression. - Robbins, Cotran & Kumar, p. 303
IV. Epigenetic Contributions
Beyond DNA mutations, epigenetic aberrations also contribute:
- DNA methylation (especially at CpG islands of tumor suppressor promoters) → gene silencing
- Histone modifications → alter chromatin accessibility → activate oncogenes or silence suppressors
- Cancer cells have abnormal methylation patterns - global hypomethylation (genomic instability) + focal hypermethylation (silencing tumor suppressors)
V. Genomic Instability - The Enabling Hallmark
Loss-of-function mutations in genes that maintain genomic integrity are often early events in solid tumor development:
- Driver mutations: directly contribute to malignant phenotype
- Passenger mutations: neutral, accumulated due to mutator phenotype (hundreds to thousands in carcinogen-induced tumors)
- By clinical detection (~10⁹ cells, ≥30 doublings), most tumors are genetically heterogeneous across subclones
- Tumor subclones undergo Darwinian selection - the fittest subclones dominate, explaining tumor progression and therapy resistance
VI. Tumor Initiation and Promotion (Chemical Carcinogenesis Model)
- Initiation: permanent DNA damage (mutation) from carcinogenic agent - irreversible
- Promotion: stimulation of initiated cell proliferation by promoters (not mutagenic alone); drives clonal expansion of mutated cells → further mutations → fully malignant clone
- Promoters are reversible (removing them stops progression at early stages)
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 251-303; Robbins & Kumar Basic Pathology, pp. 220-240
Mechanism of metastasis
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
I now have comprehensive, detailed content covering the complete molecular mechanism of metastasis. Here is the full answer:
Mechanism of Metastasis
The metastatic cascade is the sequential series of events through which a cancer cell leaves its primary site, survives in transit, and establishes a new colony at a distant organ. It is divided into two major phases: (1) invasion of the ECM and (2) vascular dissemination, tissue homing, and colonization. - Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 290
Fig. 7.36 - The metastatic cascade: sequential steps in hematogenous spread.
Why is Metastasis so Inefficient?
Although many locally invasive cells enter the bloodstream each day, very few produce actual metastases. There are no individual "metastasis genes" - the metastatic phenotype requires accumulation of multiple complementary genetic and epigenetic alterations. The "skills" needed may be present only in rare tumor subclones, or may require collaboration between subclones - some evidence even suggests successful metastases arise from cells migrating as cohesive groups.
PHASE 1: Invasion of the Extracellular Matrix
Normal tissue compartments are separated by two types of ECM:
- Basement membrane - surrounds epithelium and vascular endothelium
- Interstitial connective tissue - fills spaces between cells
For a carcinoma cell to metastasize, it must breach the basement membrane, traverse interstitial connective tissue, then penetrate the vascular basement membrane to enter circulation. This process is repeated in reverse when colonizing a distant site.
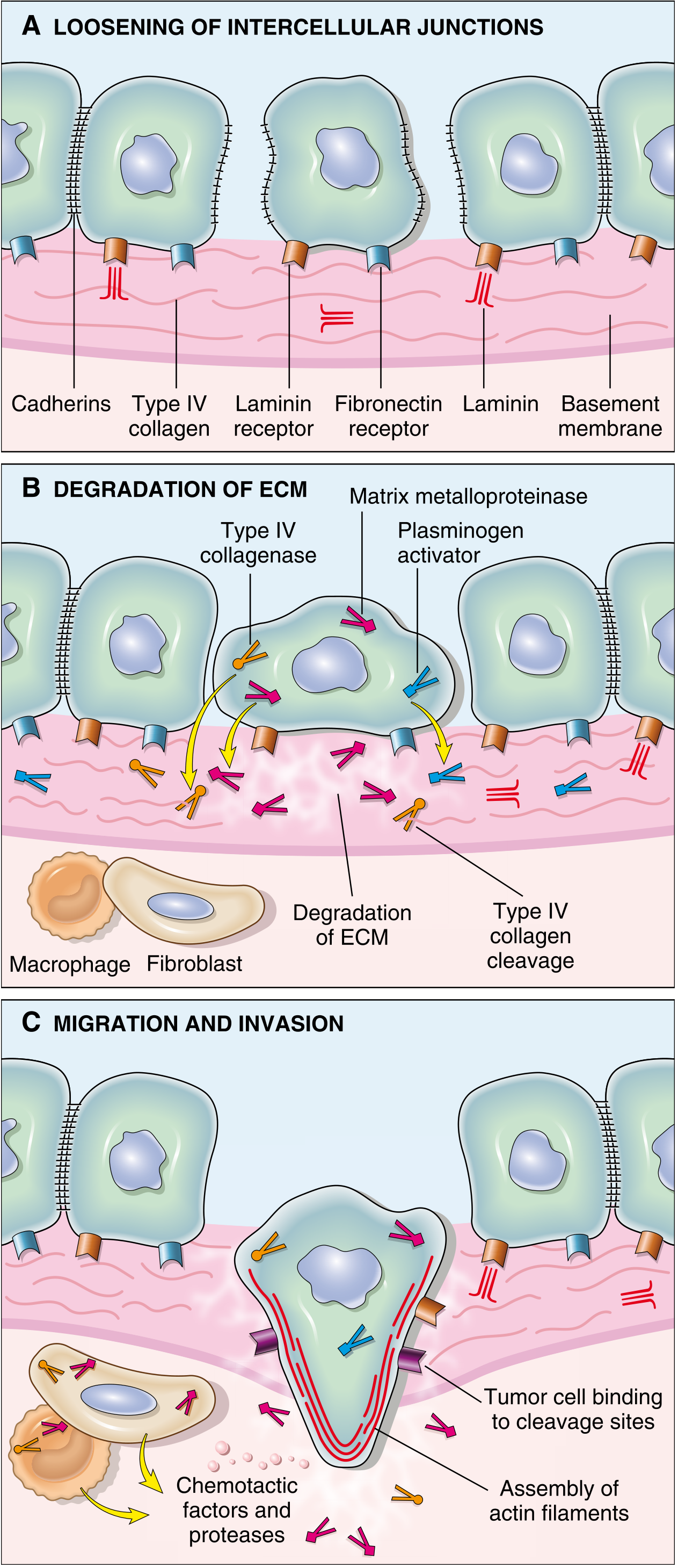
Fig. 7.37 - (A) Loosening of intercellular junctions via cadherin loss; (B) Degradation of ECM by MMPs and plasminogen activators; (C) Migration and invasion guided by chemotactic factors with actin cytoskeleton assembly.
ECM invasion proceeds through four sequential steps:
Step 1 - Loosening of Tumor Cell-Tumor Cell Interactions
Normal epithelial cells are tightly bound to each other and to the ECM by adhesion molecules, principally E-cadherin - a transmembrane glycoprotein that mediates homotypic adhesion between epithelial cells.
Mechanisms of E-cadherin loss:
- Direct mutation - loss-of-function mutations in CDH1 gene (e.g., gastric adenocarcinomas, lobular breast carcinoma)
- Epithelial-Mesenchymal Transition (EMT) - E-cadherin is transcriptionally silenced, at least transiently
EMT in detail:
- Controlled by transcription factors SNAIL and TWIST
- Not just E-cadherin loss - a full phenotypic reprogramming:
| Feature | Epithelial (original) | Mesenchymal (after EMT) |
|---|---|---|
| E-cadherin | High | Downregulated |
| Vimentin | Low | Upregulated |
| Smooth muscle actin | Low | Upregulated |
| Cell polarity | Present | Lost |
| Migration | Restricted | Promigratory |
EMT is particularly implicated in metastasis of breast and prostate carcinomas. The resulting mesenchymal phenotype confers the motility and invasiveness needed for subsequent steps.
Step 2 - Degradation of the Basement Membrane and ECM
Tumor cells degrade the basement membrane and interstitial connective tissue either by secreting proteolytic enzymes directly or by inducing stromal cells (fibroblasts, macrophages) to produce them.
Key proteolytic enzymes:
| Enzyme | Type | Role |
|---|---|---|
| MMP9 (gelatinase B) | Matrix metalloproteinase | Cleaves type IV collagen in basement membrane; also releases VEGF from ECM-sequestered pools; generates chemotactic and angiogenic fragments |
| MMP2 (gelatinase A) | Matrix metalloproteinase | Cleaves collagen IV and laminin → generates novel binding sites on tumor cells |
| Cathepsin D | Lysosomal protease | Broad ECM degradation |
| Urokinase plasminogen activator (uPA) | Serine protease | Converts plasminogen → plasmin → activates other MMPs |
Important balance: Normal tissues have metalloproteinase inhibitors (TIMPs). In malignancy, MMP activity is increased AND TIMP concentrations are reduced - double shift toward degradation.
Key point: Benign tumors of breast, colon, and stomach have little MMP9 activity; their malignant counterparts overexpress it.
Secondary effects of ECM degradation (beyond just structural breach):
- Release of VEGF sequestered in ECM → promotes angiogenesis
- Generation of collagen/proteoglycan cleavage products with chemotactic, angiogenic, and growth-promoting effects
- Creation of novel ECM binding sites that draw tumor cells forward (Step 3)
Step 3 - Attachment to Remodeled ECM Components
Tumor cells show complex changes in integrin expression (transmembrane proteins that anchor cells to ECM):
- In normal epithelial cells: integrins binding basement membrane laminin and collagens are restricted to the basal surface → keeps cells resting and polarized
- Loss of ECM adhesion normally triggers anoikis (apoptosis from loss of anchorage; literally "without a home")
- Tumor cells resist anoikis by expressing alternative integrins that transmit survival signals despite ECM detachment
Additionally, MMP2 and MMP9 cleavage of collagen IV and laminin generates novel binding sites that engage tumor cell receptors and actively stimulate migration.
Step 4 - Migration and Invasion
Locomotion is the final step that propels tumor cells through the degraded basement membrane and zones of matrix proteolysis. Migration is a cyclic ratchet process:
Attach at leading edge → detach at trailing edge → contract actin cytoskeleton → move forward
Stimuli that drive tumor cell migration:
- Tumor cell-derived autocrine motility factors - chemokines and growth factors (e.g., insulin-like growth factors, IGF)
- ECM cleavage products - collagen and laminin fragments acting as chemotactic gradients
- Stromal paracrine factors - most importantly hepatocyte growth factor/scatter factor (HGF/SF), which binds the receptor tyrosine kinase MET on tumor cells → potent stimulation of motility
Cancer-associated fibroblasts (CAFs): Under the influence of invading cancer cells, stromal fibroblasts are reprogrammed ("cancer-associated fibroblasts") and alter their expression of ECM molecules, proteases, protease inhibitors, and growth factors to further support invasion.
This phase culminates in intravasation - penetration through the endothelial basement membrane and transmigration into the vascular lumen.
PHASE 2: Vascular Dissemination, Homing, and Colonization
Survival in Circulation
Once in the bloodstream, tumor cells face multiple threats:
- Mechanical shear stress
- Anoikis (apoptosis from loss of anchorage)
- Innate and adaptive immune destruction
How cells survive:
- Tumor cell emboli/aggregates are far more likely to establish metastases than single cells
- Platelet coating - tumor cells associate with platelets via homotypic and heterotypic interactions; platelet aggregates stabilize emboli and enhance survival
- Tumor cells may express anionic substances (e.g., polyphosphate) that activate factor XII → fibrin deposition → further stabilizes emboli
- Circulating as a group provides collective competence - collectively more likely to possess all properties needed for metastasis, including stem cell-like properties for plasticity in a new microenvironment
Organ Tropism - Where Metastases Settle
Three factors determine where circulating tumor cells arrest and grow:
1. Anatomy and vascular drainage (first-pass rule)
- Portal venous drainage → liver (colon cancer)
- Systemic venous drainage → lungs
- Paravertebral venous plexus (Batson's plexus) → spine (prostate, thyroid)
2. Tumor cell tropism for specific tissues (organ-specific homing)
Despite simple anatomy predicting first-pass organs, many cancers show non-anatomic preferences:
| Tumor | Preferential Site | Mechanism |
|---|---|---|
| Prostate carcinoma | Bone | Organ-specific adhesion/growth factors |
| Breast carcinoma | Bone | PTHRP → RANKL → osteoclast activation |
| Bronchogenic carcinoma | Adrenal glands, brain | Organ-specific tropism |
| Neuroblastoma | Liver, bones | Organ-specific tropism |
| Uveal melanoma | Liver | Remarkable tropism |
Molecular mechanisms of organ tropism:
- Adhesion molecules - tumor cells express adhesion molecules (e.g., CD44) whose ligands are preferentially expressed on endothelial cells of target organs. CD44 normally guides T lymphocytes to lymphoid tissues by binding hyaluronate on high endothelial venules; solid tumors express CD44 to exploit the same mechanism
- Chemokine receptors - some cancer cells express chemokine receptors (e.g., CXCR4) that guide them toward tissues expressing the matching chemokine (CXCL12), mimicking how immune cells home to tissues
- Seed-soil hypothesis (Paget) - certain tissues provide a favorable "soil" for specific tumor "seeds"; tissues with high vascularity but rare metastases (skeletal muscle, spleen) represent "unfavorable soil"
Extravasation
Once arrested at a distant capillary bed:
- Tumor cells undergo transmigration between endothelial cells + egress through basement membrane
- Requires: integrins, laminin receptors, proteolytic enzymes, and chemokines (from tumor cells and innate immune cells like monocytes and neutrophils)
- Mechanisms differ based on endothelium type: fenestrated endothelium (liver, bone marrow) vs. tight-junction endothelium (brain)
Colonization and Tumor Dormancy
Even after successful extravasation, cells may fail to grow - this is called tumor dormancy (well described in melanoma, breast, and prostate cancer).
Productive colonization involves a feedback loop between tumor cells and the resident stroma:
Tumor cells secrete cytokines, growth factors, and ECM molecules → modify resident stromal cells → stromal cells make the metastatic site habitable ("pre-metastatic niche")
Classic example - Breast cancer → bone:
- Breast cancer cells secrete PTHrP (parathyroid hormone-related protein)
- PTHrP stimulates osteoblasts to produce RANK ligand (RANKL)
- RANKL activates osteoclasts → bone matrix degradation
- Bone matrix releases embedded growth factors: IGF, TGF-β
- IGF and TGF-β bind receptors on cancer cells → activate survival/proliferation signaling
This creates a self-amplifying feedback loop (vicious cycle of bone metastasis).
Summary: The Metastatic Cascade at a Glance
Primary tumor growth → angiogenesis
↓
Selection of metastatic subclone (EMT: SNAIL/TWIST activation, E-cadherin loss)
↓
STEP 1: Loosening of cell-cell contacts (E-cadherin loss / EMT)
↓
STEP 2: ECM degradation (MMP2, MMP9, uPA, cathepsin D)
↓
STEP 3: Attachment to remodeled ECM (altered integrins, anoikis resistance)
↓
STEP 4: Migration (autocrine IGF, HGF/MET, actin cytoskeleton ratcheting)
↓
INTRAVASATION (penetrate vascular basement membrane)
↓
CIRCULATION (platelet coating, fibrin stabilization of tumor emboli)
↓
Immune evasion in bloodstream
↓
ARREST in capillary bed (organ-specific adhesion: CD44, CXCR4)
↓
EXTRAVASATION (integrins, proteases, chemokines)
↓
COLONIZATION (stromal remodeling, pre-metastatic niche, PTHrP-RANKL-osteoclast axis)
↓
Angiogenesis + Growth = METASTATIC TUMOR
Key Concepts Summary (Robbins, Cotran & Kumar, p. 292):
- Invasion occurs in 4 steps: loosening of cell-cell contacts → ECM degradation → attachment to remodeled ECM → migration
- E-cadherin inactivation (mutations or EMT via SNAIL/TWIST) is the first step
- MMPs and cathepsins mediate ECM degradation; also release pro-tumorigenic growth factors and generate chemotactic fragments
- Metastatic site is predicted by vascular drainage (first-pass) AND organ-specific tropism
- CD44 and chemokine receptors (CXCR4) mediate organ-specific homing
- EMT transcription factors SNAIL and TWIST are the master regulators of the invasive phenotype
Source: Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 290-293
This is a shared conversation. Sign in to Orris to start your own chat.