Also explain healing and scarring mechanism in easy language as per Robbins and Cotran pathology
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Loading Image
Searching Images
wound healing scar formation steps granulation tissue collagen
I have all the content I need from Robbins & Cotran. Now let me compile a comprehensive, easy-to-understand explanation.
Healing and Scarring — Robbins & Cotran Pathology (Explained Simply)
The Big Picture First
When tissue is injured, the body tries to fix it in two ways:
| Regeneration | Scar Formation | |
|---|---|---|
| What happens | Normal cells come back | Fibrous connective tissue fills the gap |
| End result | Tissue looks and works like before | Tissue is structurally stable but not normal |
| When | Mild/superficial injury | Severe injury with damage to connective tissue framework |
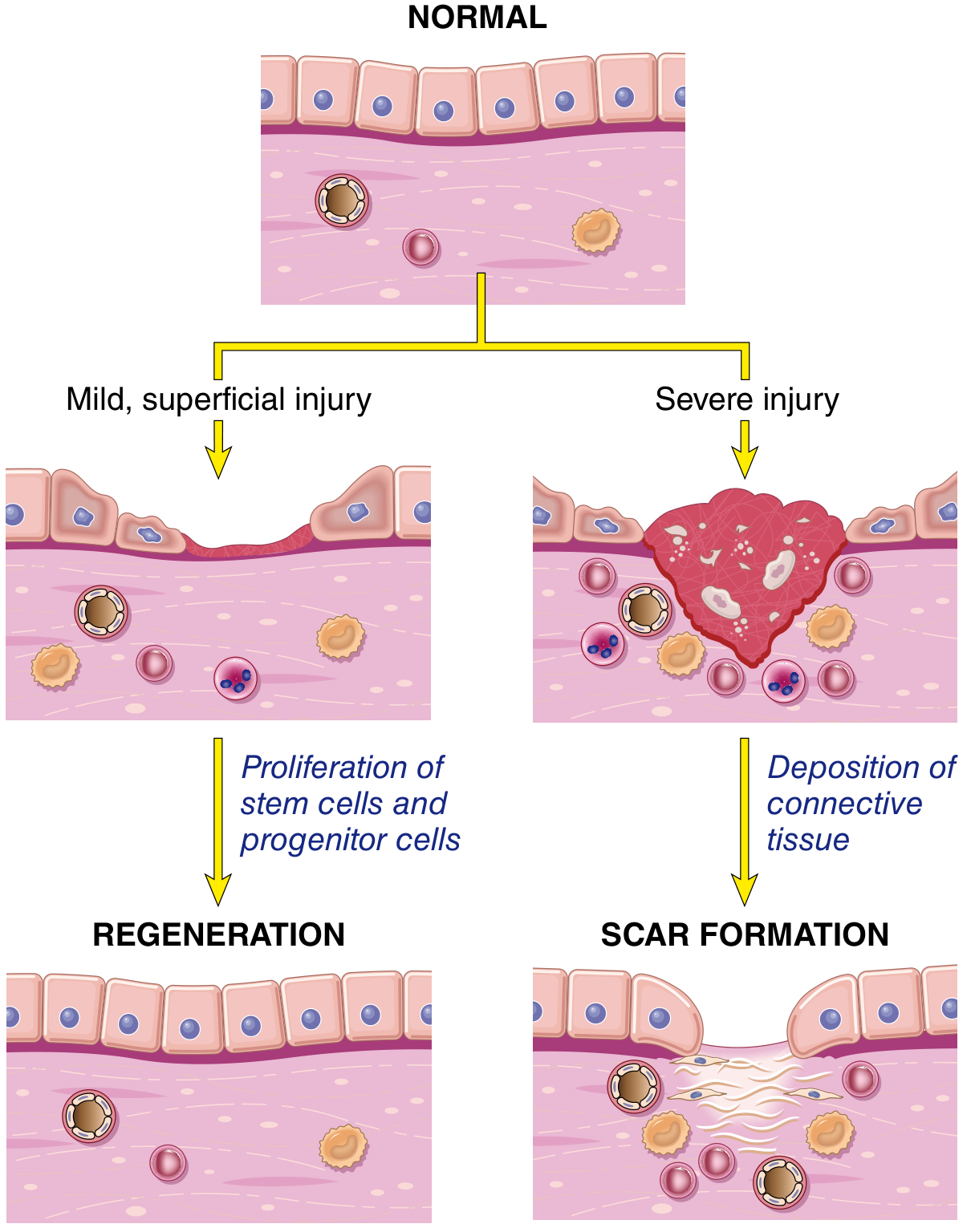
Mild injury → stem cells and progenitor cells proliferate → regeneration. Severe injury → connective tissue is deposited → scar.
Part 1: Cell Proliferation Capacity (Who Can Regenerate?)
Not all cells can regenerate equally. Robbins divides body cells into three types:
🟢 Labile (Continuously Dividing) Cells
- Always dividing, always replacing themselves
- Examples: skin epithelium, gut lining, bone marrow blood cells, oral/esophageal/cervical epithelia
- These regenerate very easily after injury
🟡 Stable (Quiescent) Cells
- Normally dormant (not dividing), but can divide when needed
- Examples: liver cells (hepatocytes), kidney tubular cells, smooth muscle, endothelial cells, fibroblasts
- They wake up and proliferate when injured — this is why the liver can regenerate after partial removal
🔴 Permanent (Non-Dividing) Cells
- Lost forever once destroyed — they cannot divide
- Examples: neurons, cardiac muscle cells (cardiomyocytes), skeletal muscle cells
- Injury here almost always leads to scarring, not regeneration
Part 2: What Drives Regeneration? (Growth Factors & ECM)
Regeneration is driven by growth factors acting on surviving cells:
| Growth Factor | Key Role |
|---|---|
| EGF / TGF-α | Stimulate proliferation of epithelial, hepatic, and other cells |
| HGF (Hepatocyte Growth Factor) | Promotes liver and epithelial cell growth, also called "scatter factor" |
| VEGF | Stimulates new blood vessel formation (angiogenesis) |
| PDGF | Recruits fibroblasts and smooth muscle cells; drives scarring |
| FGF | Angiogenesis + fibroblast activation |
| TGF-β | The key driver of fibrosis — stimulates collagen production |
The ECM (extracellular matrix) — the scaffold of proteins and fibres surrounding cells — is equally important. It:
- Provides a "highway" for migrating cells
- Stores growth factors, releasing them when tissue is damaged
- Signals cells when to stop or start dividing
Think of ECM as the building's scaffolding — without it, even cells that want to regenerate don't know where to go.
Part 3: Repair by Scarring — Steps in Scar Formation
When regeneration isn't enough (severe injury, permanent cells lost, ECM destroyed), the body fills the gap with a fibrous scar. This happens in four sequential but overlapping steps:
Step 1 — Angiogenesis (New Blood Vessels Grow In)
- New capillaries sprout into the wound
- Driven by VEGF and FGF
- These vessels are leaky and bring nutrients and inflammatory cells to the repair zone
Step 2 — Migration and Proliferation of Fibroblasts
- Fibroblasts are connective tissue cells that produce collagen
- They are recruited from the wound margins and surrounding tissue by PDGF and FGF
- They migrate into the wound and start multiplying
- Macrophages are crucial here — they secrete growth factors (PDGF, TGF-β, FGF) that keep the process going
Step 3 — Granulation Tissue Formation
Granulation tissue = new capillaries + proliferating fibroblasts + macrophages + loose ECM
This is the hallmark of repair. It:
- Appears as soft, pink, granular tissue (hence the name)
- Fills the wound space
- Is very vascular (bleeds easily when touched)
- Is the "raw material" from which the scar is built
Macrophages in granulation tissue are essential — they clear debris AND secrete the signals that drive further healing.
Step 4 — Remodelling (Scar Maturation)
- Fibroblasts lay down collagen (mainly Type III early → replaced by Type I later)
- Blood vessels regress (the scar becomes avascular and pale)
- Myofibroblasts (fibroblasts with contractile properties, like muscle) cause wound contraction — this physically pulls the wound edges together
- The scar shrinks and hardens over weeks to months
- Net result: a pale, firm, avascular scar
Think of it like wet cement drying — first soft and pliable, then it hardens and shrinks.
Part 4: Healing of Skin Wounds
Two classic patterns:
Primary Intention (Clean Surgical Wound — edges close together)
- Clot forms between the edges
- Inflammatory cells clean up debris (24–48 hrs)
- Epithelial cells migrate across the narrow gap within 24–48 hrs
- Granulation tissue forms by day 3–5
- Scar forms — small and neat
- Wound strength reaches ~70–80% of normal by 3 months
Secondary Intention (Large wound — edges far apart)
- Much more granulation tissue needed to fill the gap
- Wound contraction by myofibroblasts is very prominent (can reduce wound size by up to 80%)
- Larger, more unsightly scar
- Takes much longer to complete
Part 5: Factors That Affect Healing
Local Factors
| Factor | Effect |
|---|---|
| Infection | Most common cause of delayed healing — bacteria prolong inflammation and destroy tissue |
| Blood supply | Poor circulation → poor oxygen delivery → slow healing (e.g., diabetic foot) |
| Foreign bodies | Sutures, fragments — keep inflammation going |
| Size and location | Small, well-vascularised wounds heal better |
| Mechanical stress | Movement at wound site disrupts healing |
Systemic Factors
| Factor | Effect |
|---|---|
| Diabetes mellitus | Impaired leukocyte function, poor vascularisation, peripheral neuropathy |
| Nutritional deficiency | Vitamin C → collagen synthesis fails; zinc deficiency → impaired cell proliferation |
| Glucocorticoids (steroids) | Suppress inflammation AND collagen synthesis |
| Age | Decreased regenerative capacity in elderly |
| Haematological disorders | Anaemia, clotting disorders |
Part 6: Abnormalities in Healing
Chronic Wounds (Defects in Healing)
Occur when the normal healing process stalls, usually due to:
- Persistent infection
- Ischaemia (poor blood supply)
- Diabetes
- Sustained pressure (pressure sores)
Healing never completes → ulcer formation.
Excessive Scarring (Too Much of a Good Thing)
| Type | What Happens |
|---|---|
| Hypertrophic Scar | Excess collagen in the wound but stays within wound boundaries; can regress over time |
| Keloid | Scar tissue grows beyond the wound margins into surrounding normal skin; more common in dark-skinned individuals; does NOT regress |
| Contracture | Excessive wound contraction → deformity; serious problem in burns across joints |
| Fibrosis | Pathological scarring of organs (lung, liver, kidney) driven by chronic inflammation and persistent TGF-β signalling |
Summary Flow Chart (Simplified)
INJURY
│
├─── Mild (epithelium only)
│ └── Stem cells proliferate → REGENERATION → Normal tissue
│
└─── Severe (connective tissue damaged)
└── Angiogenesis → Fibroblast recruitment
└── GRANULATION TISSUE forms
└── Collagen deposited, vessels regress, myofibroblasts contract
└── MATURE SCAR (pale, firm, avascular)
Source: Robbins, Cotran & Kumar — Pathologic Basis of Disease, Chapter 3: Tissue Repair, pp. 109–120; and Robbins & Kumar Basic Pathology, corresponding section.
detailed outline on anemia, and its types. like hereditary spherocytosis, G6PD deficiency etc, for the level of MS1, w.r.t. Robbins & Cotran, Guyton & Hall
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Searching Images
anemia classification RBC morphology peripheral smear hereditary spherocytosis sickle cell
Finding Sources
Searching Images
sickle cell anemia peripheral smear sickling hemoglobin S
Searching Images
hereditary spherocytosis peripheral smear small spherical red cells spleen
Here's your complete MS1-level anemia outline drawn directly from Robbins, Cotran & Kumar and Guyton & Hall:
Anemia — Detailed MS1 Outline
Robbins, Cotran & Kumar — Pathologic Basis of Disease | Guyton & Hall — Medical Physiology
SECTION 1: DEFINITION & OVERVIEW
Anemia = reduction of the total circulating red cell mass below normal limits → decreased oxygen-carrying capacity → tissue hypoxia.
Diagnosed clinically by:
- ↓ Hematocrit (packed RBC volume / total blood volume)
- ↓ Hemoglobin concentration (g/dL)
"Anemia means deficiency of hemoglobin in the blood, which can be caused by too few RBCs or too little hemoglobin in the cells." — Guyton & Hall
SECTION 2: CLASSIFICATION
2A. By Mechanism (Robbins — Table 14.1)
| Mechanism | Examples |
|---|---|
| Blood loss | Acute (trauma), Chronic (GI bleed, menorrhagia) |
| ↑ Destruction (Hemolysis) | Spherocytosis, G6PD, Sickle cell, Thalassemia, PNH, Autoimmune HA |
| ↓ Production | Iron deficiency, Megaloblastic, Aplastic, Anemia of chronic disease |
2B. By Morphology (Peripheral Smear — Clinically Useful)
| Morphology | MCV | Causes |
|---|---|---|
| Microcytic hypochromic | < 80 fL | Iron deficiency, Thalassemia, Anemia of chronic disease |
| Normocytic normochromic | 80–100 fL | Acute blood loss, Aplastic anemia, Hemolysis, CKD |
| Macrocytic | > 100 fL | Megaloblastic (B12/folate deficiency), Liver disease, Hypothyroidism |
Key Red Cell Indices
| Index | Normal | What it Measures |
|---|---|---|
| MCV | 80–100 fL | Average RBC size |
| MCH | 27–33 pg | Hemoglobin per RBC |
| MCHC | 32–36 g/dL | Hemoglobin concentration in RBCs |
| RDW | < 14.5% | Variation in RBC size (↑ in iron deficiency) |
SECTION 3: HEMOLYTIC ANEMIAS (Increased Destruction)
Key concept: Hemolysis = RBC destruction before normal 120-day lifespan. Can be intravascular (within blood vessels) or extravascular (in spleen/liver macrophages — most common).
Lab hallmarks of hemolysis:
- ↓ Hb, ↓ Hematocrit
- ↑ Reticulocytes (compensatory BM response)
- ↑ Unconjugated (indirect) bilirubin → jaundice
- ↑ LDH (cell lysis marker)
- ↓ Haptoglobin (binds free Hb — gets consumed)
- Splenomegaly (from RBC trapping)
3A. HEREDITARY SPHEROCYTOSIS (HS)
Category: Inherited red cell membrane disorder
Pathogenesis
- Autosomal dominant (AD) in most; some AR
- Mutations in RBC cytoskeletal proteins:
- Ankyrin (most common), Spectrin (α or β), Band 3, Protein 4.2
- These proteins anchor the lipid bilayer to the underlying cytoskeleton
- Deficiency → membrane instability → RBC loses membrane fragments during repeated splenic passage (vesiculation)
- Surface area ↓ relative to volume → sphere shape (spherocyte) instead of biconcave disc
- Spherocytes are rigid → trapped in splenic sinusoids → destroyed by macrophages = extravascular hemolysis
"The RBCs are very small and spherical rather than biconcave discs. They cannot withstand compression forces because they do not have the normal loose, baglike membrane structure." — Guyton & Hall
Peripheral Smear
- Small, round, dark-staining spherocytes with no central pallor
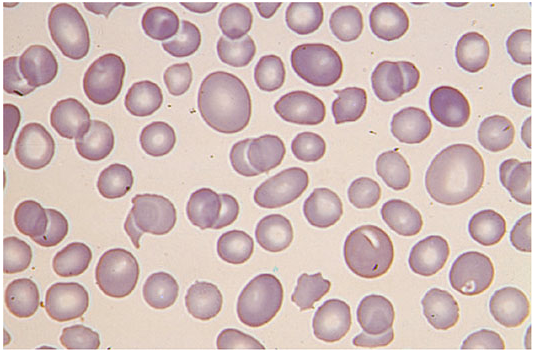
Diagnosis
- Osmotic fragility test — spherocytes lyse in hypotonic saline more readily (↑ fragility)
- EMA binding test (flow cytometry) — modern, more sensitive
- Negative direct Coombs test (distinguishes from autoimmune HA)
Clinical Features
- Anemia (variable severity), Jaundice, Splenomegaly
- Pigment gallstones (excess bilirubin → calcium bilirubinate stones)
- Aplastic crisis — triggered by Parvovirus B19 (kills erythroid precursors → sudden severe anemia)
- Megaloblastic crisis — folate demand exceeds supply
Treatment
- Splenectomy — removes site of destruction → cures anemia (spherocytes persist on smear, but no longer destroyed)
- Folic acid supplementation
3B. G6PD DEFICIENCY
Category: Inherited enzyme defect (hexose monophosphate / pentose phosphate shunt)
Pathogenesis
- X-linked recessive → mainly affects males
- G6PD = first enzyme of the HMP shunt → generates NADPH → keeps glutathione (GSH) in reduced form
- GSH = RBC's primary antioxidant (neutralizes H₂O₂ and superoxide)
- Without G6PD → NADPH depleted → GSH depleted → oxidative stress unchecked
- Oxidized hemoglobin denatures → forms Heinz bodies (precipitates on RBC membrane)
- Macrophages "bite out" Heinz bodies → bite cells / blister cells on smear
- Eventually → episodic hemolysis triggered by oxidative stressors
Triggers
| Category | Examples |
|---|---|
| Drugs | Primaquine, dapsone, sulfonamides, nitrofurantoin |
| Infections | Any febrile illness |
| Foods | Fava beans (favism — esp. Mediterranean variant) |
| Metabolic | DKA (acidosis) |
Key Variants
| Variant | Population | Severity |
|---|---|---|
| G6PD A− | African Americans | Mild — only old RBCs affected (young RBCs still have enzyme) |
| G6PD Mediterranean | Mediterranean, Middle East | Severe — all RBCs affected |
Peripheral Smear During Crisis
- Bite cells / blister cells
- Heinz bodies (seen with crystal violet stain, not standard H&E)
- Polychromasia (reticulocytosis)
Diagnosis
- G6PD enzyme assay — best done after crisis (during crisis, deficient old cells are destroyed; only young cells with higher enzyme remain → may give false-normal result)
Clinical Features
- Between episodes: Completely normal
- During episode: Acute hemolytic anemia, dark urine (hemoglobinuria), jaundice, back/abdominal pain
- Self-limiting in G6PD A− (new young RBCs replace old ones); more severe in Mediterranean variant
Treatment
- Avoid triggers (primary prevention)
- Supportive care during crisis; transfusion if severe
3C. SICKLE CELL DISEASE (SCD)
Category: Hemoglobinopathy (structurally abnormal globin)
Molecular Basis
- Autosomal recessive (codominant)
- Single point mutation in β-globin gene (chromosome 11): → Position 6: Glutamic acid → Valine → Normal HbA (α₂β₂) → HbS (α₂β^S₂)
- HbSS = sickle cell disease (severe)
- HbAS = sickle cell trait (asymptomatic, protective against P. falciparum malaria)
Mechanism of Sickling
- Deoxygenation → HbS polymerizes into long rigid tactoids (fibers)
- RBC distorts into a sickle/crescent shape
- Initially reversible (re-oxygenation un-sickles); repeated episodes → irreversibly sickled cells (permanent membrane damage)
- Sickled cells:
- Are rigid → obstruct microvessels → vaso-occlusion → ischemia/infarction
- Are fragile → destroyed by spleen → hemolysis
Factors that promote sickling: ↓ O₂ tension, dehydration, acidosis, cold, infection, high altitude
"The precipitated hemoglobin damages the cell membrane, so the cells become highly fragile, leading to serious anemia." — Guyton & Hall
Clinical Features
From Hemolysis:
- Chronic hemolytic anemia (Hb 6–9 g/dL), jaundice, gallstones, splenomegaly (early)
- Aplastic crisis (Parvovirus B19)
From Vaso-occlusion (Ischemia/Infarction):
| Organ | Manifestation |
|---|---|
| Bone/marrow | Painful vaso-occlusive crises (most common symptom — "bone pain") |
| Spleen | Autosplenectomy — repeated infarcts → fibrosed shrunken spleen → ↑ risk of encapsulated bacteria |
| Lung | Acute chest syndrome — fever, chest pain, ↓ SpO₂ — leading cause of death |
| Brain | Stroke — especially in children |
| Kidney | Renal papillary necrosis, isosthenuria (can't concentrate urine) |
| Bones | Dactylitis (hand-foot syndrome in infants), avascular necrosis of femoral head |
| Eyes | Proliferative retinopathy |
| Penis | Priapism |
| Skeleton | "H-shaped" vertebrae on X-ray (vertebral body infarcts) |
Peripheral Smear
- Sickle cells (crescent-shaped), target cells
- Howell-Jolly bodies (nuclear remnants — normally removed by spleen; present after autosplenectomy)
- ↑ Reticulocytes
Diagnosis
- Hemoglobin electrophoresis (gold standard) — HbS band present, HbA absent in HbSS
- HPLC (newborn screening)
- Sickling test (sodium metabisulfite)
Treatment
- Hydroxyurea — ↑ HbF production (HbF inhibits HbS polymerization → ↓ sickling, ↓ crises)
- Penicillin prophylaxis (for asplenic patients)
- Vaccinations (pneumococcal, meningococcal, H. flu)
- Folic acid supplementation
- Exchange transfusion (acute chest syndrome, stroke)
- Bone marrow transplantation (curative)
3D. THALASSEMIA
Category: Hemoglobinopathy — deficient globin chain synthesis
Normal Hb: α₂β₂ (HbA). Thalassemias = ↓ synthesis of one globin chain → the other accumulates → precipitates → damages RBCs.
β-Thalassemia
- Mutations in β-globin gene (chromosome 11) — mostly point mutations (>200 known)
- β⁰ = no β-chain produced; β⁺ = reduced β-chain produced
- ↓ β-globin → excess α-chains accumulate → precipitate as inclusions → damage RBC membrane → ineffective erythropoiesis (death of precursors in BM) + hemolysis
- Compensatory massive erythropoiesis → bone marrow expansion → skeletal deformities
| Genotype | Syndrome | Severity |
|---|---|---|
| β/β | Normal | — |
| β/β⁺ or β/β⁰ | β-Thalassemia Minor (Trait) | Mild microcytosis, clinically silent |
| β⁺/β⁺ | β-Thalassemia Intermedia | Moderate anemia, variable transfusion need |
| β⁰/β⁰ | β-Thalassemia Major (Cooley's anemia) | Severe, transfusion-dependent from 6 months |
β-Thalassemia Major Clinical Features:
- Severe hemolytic anemia appearing at 6 months (HbF → HbA switch)
- Massive hepatosplenomegaly (extramedullary hematopoiesis)
- "Crew-cut" skull X-ray (expanded diploe — perpendicular trabeculae)
- Chipmunk facies (maxillary bone overgrowth)
- Growth retardation
- Iron overload (from transfusions + ↑ GI absorption) → hemosiderosis → heart failure, cirrhosis, endocrine failure (the main cause of death)
- Lab: ↑ HbF, absent/↓ HbA, ↑ HbA₂
Treatment: Regular transfusions + iron chelation (deferoxamine/deferasirox); BMT (curative)
α-Thalassemia
- Deletions in α-globin genes (chromosome 16) — normal = 4 α-genes
| Deletions | Syndrome | Features |
|---|---|---|
| 1 gene deleted | Silent carrier | Normal |
| 2 genes deleted | α-Thalassemia trait | Mild microcytic anemia |
| 3 genes deleted | HbH disease | Moderate hemolytic anemia; excess β-chains → HbH (β₄) |
| 4 genes deleted | Hydrops fetalis | Incompatible with life; excess γ-chains → Hb Bart's (γ₄) — can't deliver O₂ |
3E. AUTOIMMUNE HEMOLYTIC ANEMIA (AIHA)
| Type | Antibody | Temperature | Causes |
|---|---|---|---|
| Warm AIHA | IgG | 37°C | Idiopathic, SLE, CLL, drugs (methyldopa) |
| Cold AIHA | IgM | < 30°C | Mycoplasma, EBV, idiopathic |
Diagnosis: Direct Coombs test (DAT) — positive
Treatment: Corticosteroids (warm), cold avoidance, rituximab
3F. PAROXYSMAL NOCTURNAL HEMOGLOBINURIA (PNH)
- Acquired mutation in PIG-A gene → loss of GPI-anchor proteins (CD55, CD59)
- CD55/CD59 normally protect RBCs from complement attack
- Without them → complement destroys RBCs → intravascular hemolysis
- Classic triad: hemolytic anemia + thrombosis (unusual sites: hepatic vein) + cytopenias
- Hemoglobinuria worst in morning (acidosis during sleep → complement activation)
- Diagnosis: Flow cytometry (loss of CD55/CD59)
- Treatment: Eculizumab (anti-C5 complement inhibitor)
SECTION 4: ANEMIAS OF DIMINISHED ERYTHROPOIESIS
4A. IRON DEFICIENCY ANEMIA — Most common anemia worldwide
Iron Metabolism
- Absorbed in duodenum/proximal jejunum (needs acidic pH; enhanced by vitamin C; inhibited by phytates, tea)
- Stored as ferritin (soluble) and hemosiderin (insoluble) in liver, spleen, marrow
- Transported by transferrin in plasma
Causes
| Category | Examples |
|---|---|
| Chronic blood loss (most common in adults) | GI bleeding (peptic ulcer, colorectal cancer), menorrhagia |
| Inadequate intake | Poor diet, infancy, pregnancy |
| Malabsorption | Celiac disease, post-gastrectomy, achlorhydria |
⚠️ "An alert clinician investigating unexplained iron deficiency anemia occasionally discovers an occult bleeding source such as cancer and thereby saves a life." — Robbins & Cotran
Lab Findings
| Test | Iron Deficiency Anemia |
|---|---|
| Serum iron | ↓ |
| TIBC | ↑ (body makes more transferrin to grab every iron molecule) |
| Serum ferritin | ↓↓↓ (earliest and most sensitive marker) |
| Transferrin saturation | ↓ (< 15%) |
| MCV | ↓ (microcytic) |
| MCHC | ↓ (hypochromic) |
| RDW | ↑ (high variation in cell size) |
| Reticulocytes | Low (production failure) |
Stages of Depletion
- Pre-latent: Stores depleted (↓ ferritin), normal Hb
- Latent: ↓ serum iron, ↑ TIBC, normal Hb
- IDA: Microcytic hypochromic anemia appears
Clinical Features
- Fatigue, pallor, exertional dyspnea
- Pica — craving for non-food items (ice, clay)
- Koilonychia — spoon-shaped nails
- Angular stomatitis, atrophic glossitis
- Plummer-Vinson syndrome — IDA + esophageal webs + dysphagia (→ ↑ risk esophageal carcinoma)
Treatment
- Find and treat underlying cause
- Oral ferrous sulfate for 3–6 months (continue 3 months after Hb normalizes to replenish stores)
- IV iron (malabsorption, intolerance, severe)
- Reticulocyte response in 5–10 days = confirms diagnosis
4B. MEGALOBLASTIC ANEMIA (Vitamin B12 / Folate Deficiency)
Core Concept
Both B12 and folate needed for DNA synthesis (thymidylate production). Deficiency → cells grow but cannot divide → large immature cells = megaloblasts in BM → macrocytes in blood. All rapidly dividing cells affected (including neutrophils → hypersegmented neutrophils).
"The RBCs grow too large, with odd shapes, and are called megaloblasts... the cells are mostly oversized, have bizarre shapes, and fragile membranes." — Guyton & Hall
Vitamin B12 Deficiency
Absorption pathway:
Dietary B12 (animal products) → binds Intrinsic Factor (IF) from gastric parietal cells → B12-IF complex absorbed at terminal ileum
Causes:
| Cause | Example |
|---|---|
| ↓ IF | Pernicious anemia (autoimmune — anti-parietal cell Ab, anti-IF Ab), total gastrectomy |
| ↓ Ileal absorption | Crohn's disease (terminal ileum), ileal resection, fish tapeworm (Diphyllobothrium) |
| ↓ Dietary intake | Strict vegans (body stores last 3–5 years) |
Pernicious Anemia (most important cause):
- Autoimmune destruction of parietal cells
- Anti-parietal cell antibodies (destroy the cells); anti-IF antibodies (block B12-IF binding)
- Associated with Hashimoto's thyroiditis, T1DM
- ↑ risk of gastric adenocarcinoma
Unique to B12 deficiency (not folate):
- Subacute combined degeneration of the spinal cord — degeneration of dorsal columns (vibration/proprioception loss) + lateral corticospinal tracts (upper motor neuron signs) + peripheral neuropathy
- Neurological damage progresses even if hematologic anemia is "corrected" by giving folate alone → dangerous
Folate Deficiency
Causes:
- Poor diet (alcoholics, elderly, overcooking vegetables)
- ↑ Demand (pregnancy, hemolytic anemia, rapid growth)
- Malabsorption (celiac disease)
- Drugs: Methotrexate, trimethoprim, phenytoin (folate antagonists)
⚠️ Folate in pregnancy: Deficiency → neural tube defects (spina bifida, anencephaly). Supplement from before conception.
Lab Findings (Both B12 and Folate)
| Test | Finding |
|---|---|
| MCV | > 100 fL (macrocytic) |
| Peripheral smear | Hypersegmented neutrophils (≥5 lobes or ≥1 cell with 6 lobes) — pathognomonic |
| Bone marrow | Megaloblasts, giant bands |
| Serum B12 | ↓ in B12 deficiency |
| Serum/RBC folate | ↓ in folate deficiency |
| Homocysteine | ↑ in both |
| Methylmalonic acid (MMA) | ↑ in B12 only — key differentiator |
Treatment
- B12: IM cyanocobalamin (oral ineffective in pernicious anemia/malabsorption)
- Folate: Oral folic acid
- ⚠️ Never give folate alone if B12 deficiency suspected — corrects blood picture but neurological damage continues
4C. APLASTIC ANEMIA
Definition: Hypocellular bone marrow → pancytopenia (↓ RBCs + ↓ WBCs + ↓ platelets)
Causes
| Type | Mechanism | Examples |
|---|---|---|
| Acquired (>80%) | T-cell mediated autoimmune destruction of hematopoietic stem cells | Idiopathic (50%), drugs (chloramphenicol, benzene), radiation, viral (hepatitis, EBV) |
| Inherited | Genetic defects | Fanconi's anemia (AR, DNA repair defect — short stature, radial defects, ↑ risk of AML) |
"Exposure to high-dose radiation or chemotherapy... damage stem cells of the bone marrow... insecticides or benzene in gasoline may cause the same effect." — Guyton & Hall
Lab Findings
- Pancytopenia
- Hypocellular bone marrow with fatty replacement (> 70% fat on biopsy) — diagnostic
- ↓ Reticulocytes (production failure)
- Normal RBC morphology
Clinical Features
- Anemia symptoms (fatigue, pallor)
- Infections (from neutropenia — most life-threatening)
- Bleeding (thrombocytopenia — petechiae, ecchymoses)
Treatment
- Severe + young with matched donor: Bone marrow transplantation (curative)
- Severe without donor: Immunosuppression (anti-thymocyte globulin + cyclosporine + eltrombopag)
- Supportive: transfusions, G-CSF
4D. ANEMIA OF CHRONIC DISEASE (ACD)
Most common anemia in hospitalized patients
Pathogenesis
- Chronic infection / autoimmune disease / cancer → chronic inflammation → IL-6 → ↑ hepcidin (liver-derived)
- Hepcidin degrades ferroportin (iron exporter on macrophages and gut enterocytes)
- Result: iron trapped in macrophage stores, ↓ GI iron absorption → functional iron deficiency (iron present but not accessible for erythropoiesis)
- Also: ↓ EPO response, ↓ RBC lifespan
Key Lab Comparison
| Test | ACD | Iron Deficiency |
|---|---|---|
| Serum iron | ↓ | ↓ |
| TIBC | ↓ or normal | ↑ |
| Ferritin | ↑ | ↓↓↓ |
| Transferrin saturation | ↓ | ↓ |
"Storage iron in the bone marrow and serum ferritin are increased [in ACD], in contrast to iron deficiency anemia." — Robbins & Cotran
Treatment
- Treat underlying disease
- ESAs (erythropoietin-stimulating agents) in CKD
- IV iron if also functionally iron-depleted
SECTION 5: PHYSIOLOGICAL EFFECTS OF ANEMIA — Guyton & Hall
Cardiovascular Compensation
- ↓ Blood viscosity → ↓ peripheral resistance → ↑ cardiac output (↑ HR + ↑ SV)
- Chronic severe anemia → high-output state → heart failure
- Vasodilation from tissue hypoxia-induced local mediators
O₂ Delivery Compensation
- ↑ 2,3-BPG (DPG) in RBCs → right-shifts O₂-Hb dissociation curve → Hb releases O₂ more easily to tissues
- ↑ O₂ extraction fraction per unit blood
Erythropoietin Response
- Kidney peritubular cells sense ↓ O₂ → ↑ EPO → BM → ↑ erythroid progenitor proliferation → ↑ reticulocyte release
- Reticulocyte response begins in 3–5 days, peaks at 7–10 days
Symptoms by Severity
| Hb (g/dL) | Typical Symptoms |
|---|---|
| 10–12 | Fatigue on exertion, mild pallor |
| 7–10 | Exertional dyspnea, tachycardia, palpitations |
| < 7 | Rest dyspnea, angina (if CAD), heart failure, CNS symptoms |
| < 5 | Life-threatening cardiac decompensation |
SECTION 6: MASTER COMPARISON TABLE
| Feature | Hereditary Spherocytosis | G6PD Deficiency | Sickle Cell | β-Thal Major | Iron Deficiency | B12/Folate | Aplastic |
|---|---|---|---|---|---|---|---|
| Inheritance | AD | X-linked recessive | AR | AR | Acquired | Acquired | Acquired |
| Defect | Membrane (ankyrin, spectrin) | Enzyme (HMP shunt) | Hb structure (Glu→Val) | Hb synthesis (β↓) | Iron supply | Vitamin (DNA synthesis) | Stem cells |
| MCV | Normal/slight ↓ | Normal | Normal | Microcytic ↓↓ | Microcytic ↓↓ | Macrocytic ↑↑ | Normal |
| Smear finding | Spherocytes | Bite cells, Heinz bodies | Sickle cells, targets, HJ bodies | Targets, nucleated RBCs, teardrop cells | Pencil cells, microcytes | Hyperseg. neutrophils | Normal morphology |
| Hemolysis type | Extravascular | Intra + extravascular | Both | Extravascular + ineffective erythropoiesis | None | None | None |
| Splenomegaly | ✓ | — | Early ✓ → autosplenectomy | Massive ✓ | — | — | — |
| Key diagnostic test | Osmotic fragility / EMA flow | G6PD enzyme assay | Hb electrophoresis | Hb electrophoresis | Ferritin ↓↓, TIBC ↑ | MMA ↑ (B12), homocysteine ↑ | BM biopsy (hypocellular) |
| Key treatment | Splenectomy | Avoid triggers | Hydroxyurea | Transfusion + iron chelation | Oral iron | IM B12 / oral folate | BMT / immunosuppression |
SECTION 7: MEMORY AIDS
| Topic | Mnemonic / Trick |
|---|---|
| G6PD mechanism | "No G6PD → No NADPH → No GSH → No antioxidant → Oxidative crisis → Heinz bodies → Bite cells" |
| IDA vs. ACD | Ferritin is the key: ↓ in IDA (stores empty), ↑ in ACD (stores full but locked) |
| B12 vs. Folate | Both: macrocytic anemia + hyperseg neutrophils. Only B12: neurological damage (subacute combined degeneration) + ↑ MMA |
| α-thal chromosomes | Alpha = chromosome 16 (A is the 1st letter); Beta = chromosome 11 (B is 2nd letter) |
| Sickle cell trait | HbAS = Asymptomatic + Anti-malarial (protective against P. falciparum) |
| Splenectomy in HS | Removes the "execution chamber" — spherocytes persist on smear but survive longer |
| Pernicious anemia | Parietal cells make IF; if destroyed → no IF → no B12 absorption from terminal ileum |
| Thalassemia major skeleton | "Crew-cut skull, chipmunk face, H-vertebrae" = all from marrow expansion/infarction |
Sources: Robbins, Cotran & Kumar — Pathologic Basis of Disease, Ch. 14; Robbins & Kumar Basic Pathology, Ch. 13; Guyton and Hall Textbook of Medical Physiology, Ch. 33 (Red Blood Cells, Anemia, and Polycythemia).
membrane potential w.r.t. physiology for MS level. refer Guyton & Hall
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Reading File
Loading Image
Reading File
I now have comprehensive content and the key diagrams from Guyton & Hall. Let me compile the full MS-level outline:
Membrane Potential — Physiology (MS Level)
Guyton and Hall Textbook of Medical Physiology, Chapters 5 & 6
SECTION 1: BASIC CONCEPTS — What Is a Membrane Potential?
The membrane potential is the electrical voltage difference that exists across the cell membrane at any given moment.
- Measured as: potential inside the cell relative to outside
- At rest: −70 mV in a large nerve fiber (inside is negative relative to outside)
- This negativity is not just passively sitting there — it is actively maintained by ion transport mechanisms
"The potential inside the fiber is 70 mV more negative than the potential in the extracellular fluid on the outside of the fiber." — Guyton & Hall
The membrane potential exists because:
- There are unequal ion concentrations on the two sides of the membrane
- The membrane has selective permeability — it lets some ions pass more easily than others
- The Na⁺-K⁺ pump actively maintains the concentration gradients
SECTION 2: ION CONCENTRATIONS ACROSS THE MEMBRANE
| Ion | Outside (mEq/L) | Inside (mEq/L) | Ratio |
|---|---|---|---|
| Na⁺ | 142 | 14 | ~10:1 (outside > inside) |
| K⁺ | 4 | 140 | ~35:1 (inside > outside) |
| Cl⁻ | 103 | 4 | ~26:1 (outside > inside) |
| Large anions (protein⁻, organic phosphates) | Negligible | High | Trapped inside |
These gradients are maintained by the Na⁺-K⁺ ATPase pump and selective membrane permeability.
SECTION 3: DIFFUSION POTENTIALS AND THE NERNST EQUATION
3A. Diffusion Potential — The Core Concept
When ions move across a membrane down a concentration gradient, they carry electrical charge with them. This creates a diffusion potential (voltage) that opposes further net movement of that ion.
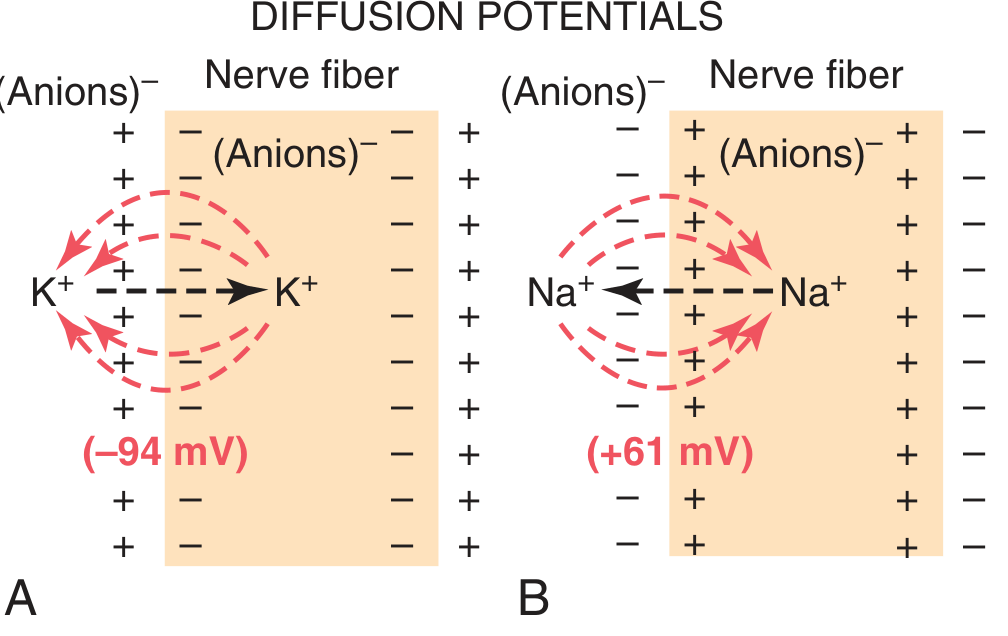
- K⁺ diffuses outward (high inside → low outside) → carries positive charges out → inside becomes negative → −94 mV (Nernst potential for K⁺)
- Na⁺ diffuses inward (high outside → low inside) → carries positive charges in → inside becomes positive → +61 mV (Nernst potential for Na⁺)
3B. The Nernst Equation
The Nernst potential for an ion is the exact membrane voltage at which the electrical force exactly balances the concentration force — so there is zero net movement of that ion.
$$\text{EMF (mV)} = \frac{\pm 61}{z} \times \log \frac{\text{[Ion]}\text{inside}}{\text{[Ion]}\text{outside}}$$
Where:
- EMF = electromotive force (the Nernst potential)
- z = electrical charge of the ion (+1 for Na⁺/K⁺, +2 for Ca²⁺, −1 for Cl⁻)
- 61 = constant at 37°C (body temperature)
Worked examples:
- K⁺: 61/1 × log(140/4) = 61 × log(35) = 61 × 1.544 = −94 mV (inside negative because K⁺ leaves)
- Na⁺: 61/1 × log(14/142) = 61 × log(0.1) = 61 × (−1) = −61 mV; but since Na⁺ enters, polarity is +61 mV inside
Remember: if the ion is positive and diffuses OUT, inside becomes negative. If it diffuses IN, inside becomes positive.
3C. The Goldman-Hodgkin-Katz (GHK) Equation
The Nernst equation handles only one ion at a time. In reality, multiple ions are permeable simultaneously. The Goldman equation accounts for relative permeabilities:
$$V_m = 61 \log \frac{P_K[K^+]o + P{Na}[Na^+]o + P{Cl}[Cl^-]_i}{P_K[K^+]i + P{Na}[Na^+]i + P{Cl}[Cl^-]_o}$$
Where P = permeability coefficient of each ion.
- At rest: membrane is ~100x more permeable to K⁺ than Na⁺ → K⁺ dominates → resting potential (~−70 mV) is closer to K⁺ Nernst potential (−94 mV) than to Na⁺ Nernst potential (+61 mV)
- During action potential: membrane becomes transiently ~500–1000x more permeable to Na⁺ → potential swings toward +61 mV → the overshoot
SECTION 4: THE Na⁺-K⁺ ATPase PUMP
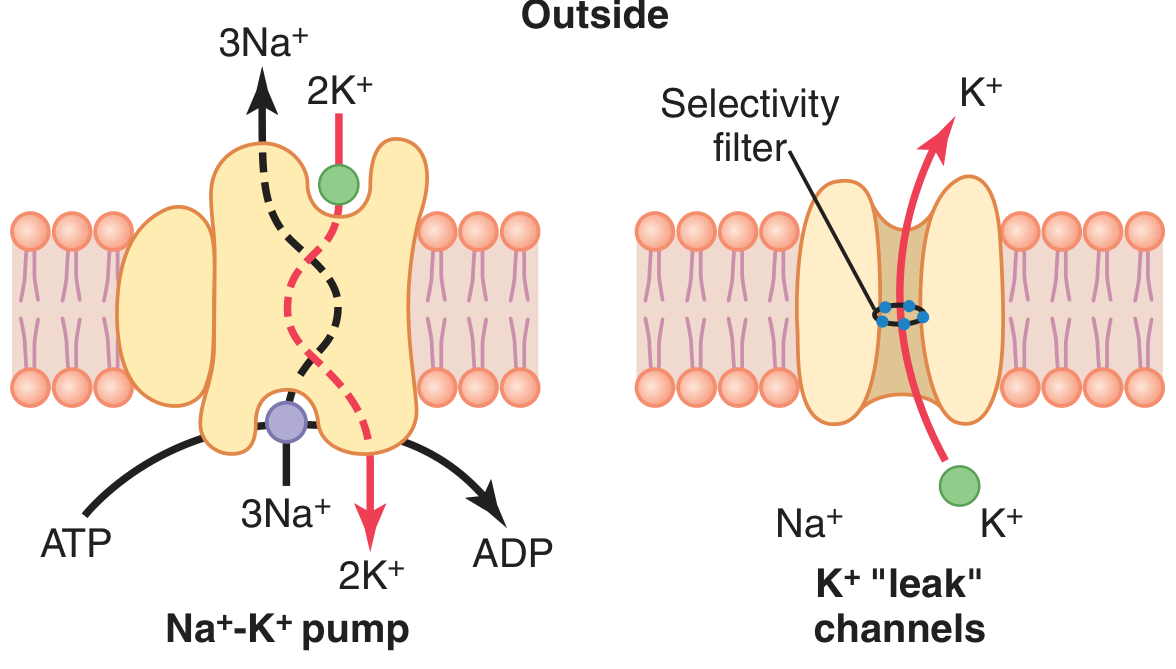
Mechanism
- Pumps 3 Na⁺ OUT and 2 K⁺ IN per ATP hydrolyzed
- Uses 1 ATP molecule per cycle
- The protein has two conformational states (E1 and E2) — binds Na⁺ intracellularly and K⁺ extracellularly
Two Roles of the Na⁺-K⁺ Pump
| Role | Details |
|---|---|
| Maintains concentration gradients | Keeps Na⁺ high outside, K⁺ high inside — the "battery" for all membrane potential activity |
| Electrogenic contribution | Pumps 3+ out for 2+ in = net loss of 1 positive charge per cycle = contributes −4 mV directly to resting potential |
"This is an electrogenic pump because three Na⁺ ions are pumped to the outside for each two K⁺ ions to the inside, leaving a net deficit of positive ions on the inside." — Guyton & Hall
K⁺ Leak Channels
- Always open at rest (also called tandem pore domain or background K⁺ channels)
- Allow K⁺ to continuously diffuse outward down its concentration gradient
- This ongoing K⁺ leak is the primary reason the resting membrane potential is negative
- Membrane is ~100x more permeable to K⁺ than Na⁺ at rest
SECTION 5: ORIGIN OF THE RESTING MEMBRANE POTENTIAL
The resting membrane potential of −70 mV arises from three contributions:
Contribution 1 — K⁺ Diffusion Potential (dominant)
- High intracellular K⁺ → K⁺ leaks out through K⁺ leak channels
- Carries positive charges out → inside becomes negative
- Alone would produce −94 mV (the K⁺ Nernst potential)
Contribution 2 — Na⁺ Leak (opposing)
- Small but constant inward Na⁺ leak through K⁺ leak channels
- Brings positive charges in → partially offsets the negative potential
- Pulls potential up from −94 mV toward zero
Contribution 3 — Na⁺-K⁺ Pump (electrogenic, minor)
- Direct electrogenic contribution of ~−4 mV
- Major role is maintaining the ion gradients, not the direct electrogenic effect
Net result:
- K⁺ contribution alone: −94 mV
- Na⁺ leak partially offsets: pulls toward +61 mV
- Final resting potential: −70 mV (a weighted average based on relative permeabilities)
Summary: resting potential = largely determined by K⁺ concentration gradient + K⁺ leak channels, slightly reduced in magnitude by background Na⁺ leak.
SECTION 6: THE ACTION POTENTIAL
6A. Definition
An action potential is a rapid, transient, self-propagating reversal of the membrane potential caused by sudden changes in ion permeability.
6B. The Action Potential Curve — Five Stages
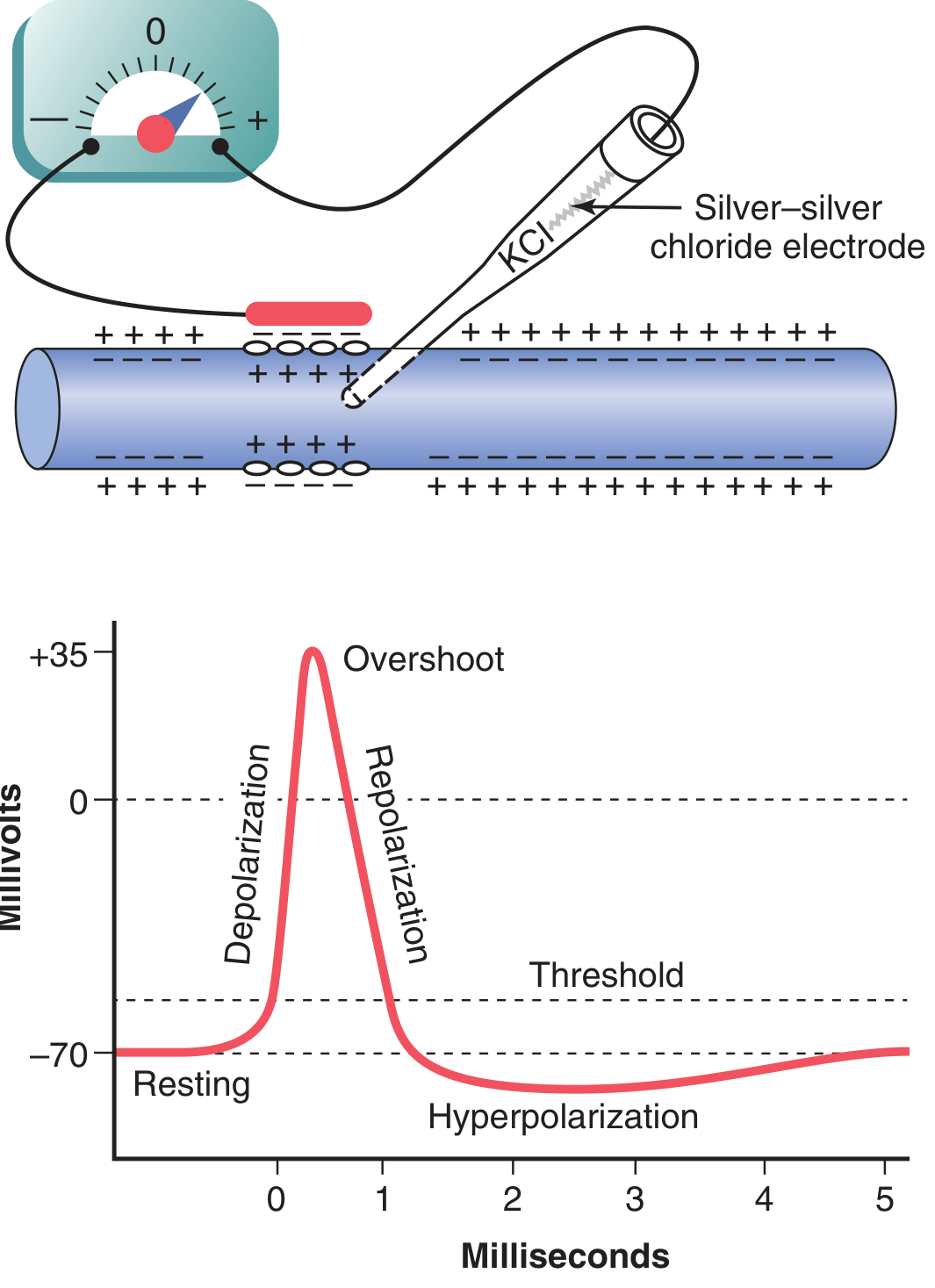
| Stage | Voltage | What Happens |
|---|---|---|
| 1. Resting | −70 mV | K⁺ leak channels open; Na⁺ channels closed; membrane "polarized" |
| 2. Depolarization | −70 → 0 mV | Stimulus → voltage-gated Na⁺ channels open → Na⁺ rushes in → positive feedback cycle |
| 3. Overshoot | 0 → +35 mV | Continued Na⁺ influx drives potential to positive values |
| 4. Repolarization | +35 → −70 mV | Na⁺ channels inactivate; voltage-gated K⁺ channels open → K⁺ rushes out |
| 5. Hyperpolarization (undershoot) | −70 → −80 mV | K⁺ channels stay open too long → excess K⁺ loss → potential briefly more negative than resting |
6C. Conductance Changes During the Action Potential
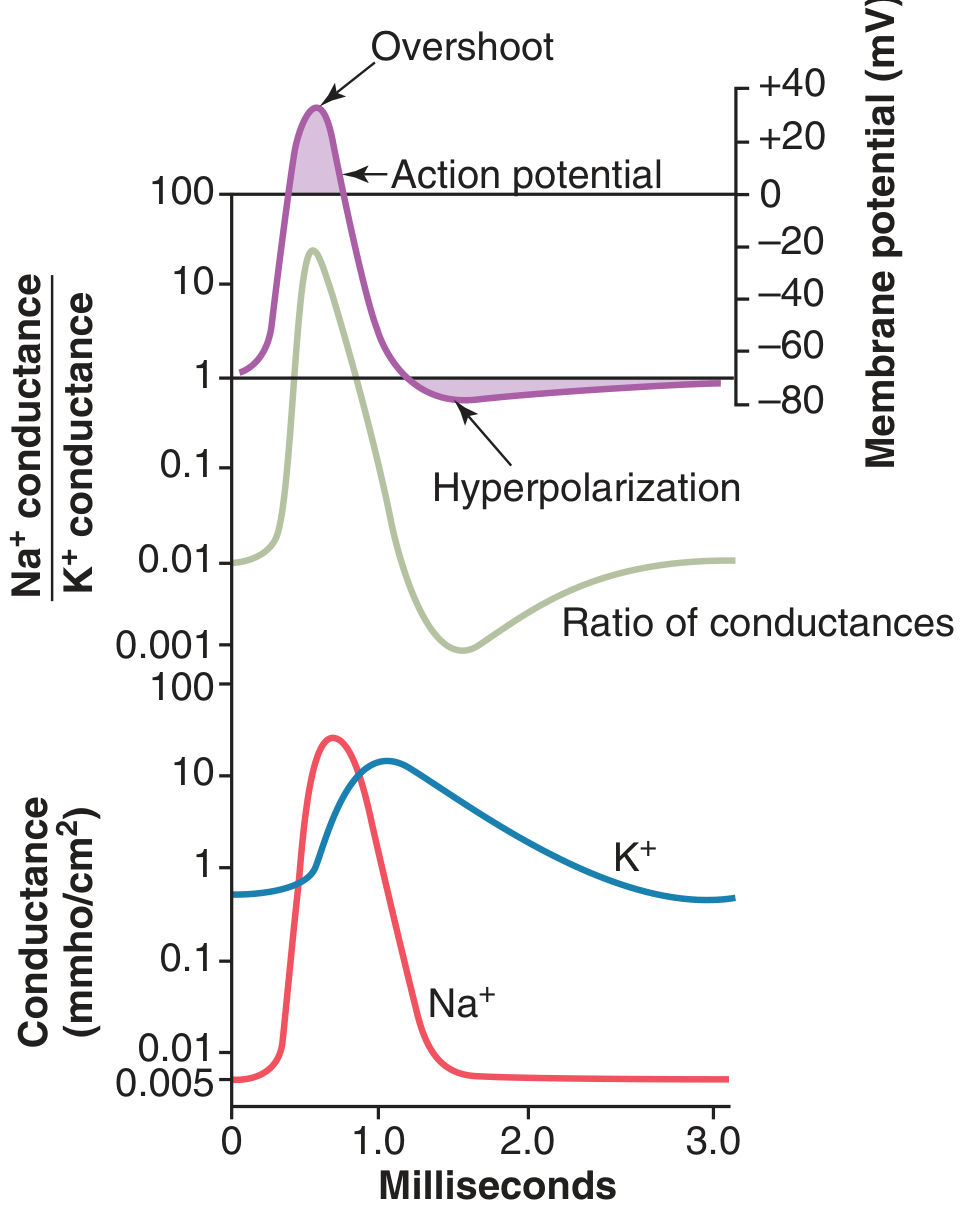
- At rest: K⁺ conductance 50–100x > Na⁺ conductance
- At depolarization onset: Na⁺ conductance increases up to 5000-fold instantaneously
- Na⁺ channels then inactivate within a fraction of a millisecond
- K⁺ channels open slowly (~0.5 ms after Na⁺ channels) and remain open longer
- Na⁺/K⁺ conductance ratio: >1000-fold increase during early action potential → drives overshoot
SECTION 7: VOLTAGE-GATED ION CHANNELS IN DETAIL
7A. Voltage-Gated Na⁺ Channel — Three States
The Na⁺ channel has two gates:
- Activation gate (m-gate): near the outer face — opens with depolarization
- Inactivation gate (h-gate): near the inner face — closes with slight delay after depolarization
| State | Activation gate | Inactivation gate | Ion Flow |
|---|---|---|---|
| Resting (closed) | Closed | Open | No Na⁺ flow |
| Activated (open) | Open | Open | Na⁺ rushes in |
| Inactivated | Open | Closed | No Na⁺ flow |
Key point: The inactivated state is different from the resting closed state. The inactivation gate must be "reset" — this requires the membrane to repolarize. This is why the channel cannot immediately be re-opened — it underlies the refractory period.
7B. Voltage-Gated K⁺ Channel — Two States (Simpler)
- Closed at rest
- Opens slowly when membrane depolarizes (delayed ~0.5 ms after Na⁺ channels)
- Closes slowly when membrane repolarizes → explains why hyperpolarization (undershoot) occurs
- Has only one gate (activation gate; no separate inactivation gate)
SECTION 8: THRESHOLD AND ALL-OR-NONE LAW
Threshold Potential
- The membrane potential must be depolarized to approximately −55 mV to trigger an action potential
- That means the membrane needs to be depolarized by ~15–20 mV from resting (from −70 to −55 mV)
- Below threshold: Graded, local, non-propagating potential (decremental)
- At/above threshold: Explosive positive feedback → full action potential fires
Positive Feedback Cycle (Hodgkin Cycle)
Depolarization → voltage-gated Na⁺ channels open → Na⁺ influx → more depolarization → more Na⁺ channels open → more Na⁺ influx... (runaway until all channels activated)
"This process is a positive-feedback cycle that, once the feedback is strong enough, continues until all the voltage-gated sodium channels have become activated." — Guyton & Hall
All-or-None Law
An action potential either fires fully or not at all — stimulus strength does not affect the size of the action potential. What changes with stimulus strength is the frequency of firing (rate coding).
SECTION 9: PROPAGATION OF THE ACTION POTENTIAL
Once an action potential fires at one spot, it spreads along the entire fiber by local circuit currents:
- Na⁺ enters at the excited zone → positive charges flow inward
- Positive charges spread laterally along the inside of the axon
- This depolarizes adjacent membrane to threshold
- New Na⁺ channels open in the adjacent zone → action potential fires there
- Process continues in both directions
Direction of propagation: Normally in one direction (away from the cell body) because the region just behind the wave front is in the refractory period and cannot be re-excited.
Propagation Velocity
| Fiber Type | Myelinated? | Velocity |
|---|---|---|
| Large myelinated (A-α) | Yes | 70–120 m/s |
| Small myelinated (A-δ) | Yes | 5–30 m/s |
| Unmyelinated (C fibers) | No | 0.5–2 m/s |
SECTION 10: SALTATORY CONDUCTION IN MYELINATED FIBERS
- Myelin sheath = layers of lipid-rich membrane wound around the axon
- Acts as an excellent electrical insulator — prevents ion leakage through internodal segments
- Nodes of Ranvier = small gaps (~1–2 µm) in the myelin sheath where Na⁺ channels are densely concentrated
- Action potential jumps from node to node (saltatory = "to jump" in Latin)
Advantages:
- Faster conduction (action potential skips long internodal segments)
- Energy efficient — Na⁺/K⁺ pump only needs to restore ions at nodes, not along the entire axon
- Allows large, fast axons in a compact space
SECTION 11: REFRACTORY PERIODS
Absolute Refractory Period (ARP)
- Lasts ~1 ms after an action potential in large nerve fibers
- During this time, no stimulus, however strong, can trigger another action potential
- Caused by: Na⁺ channel inactivation gates are closed — channels are in the inactivated state
- Cannot fire until the inactivation gate resets (requires repolarization)
Relative Refractory Period (RRP)
- Follows the ARP, lasts a few more milliseconds
- Some Na⁺ channels have recovered (reset to resting state), but K⁺ channels are still open → membrane is hyperpolarized
- A stronger-than-normal stimulus can trigger an action potential
- The resulting action potential will be slightly smaller than normal
Clinical importance: Refractory period determines the maximum frequency of nerve firing:
- Large nerve fibers: up to 1000 impulses/second
- Most CNS neurons: much lower rates due to longer refractory periods
SECTION 12: SPECIAL MEMBRANE POTENTIALS IN OTHER TISSUES
Cardiac Muscle (Brief Comparison)
| Feature | Skeletal muscle nerve | Cardiac muscle |
|---|---|---|
| Resting potential | −70 mV | −90 mV |
| Duration of AP | ~1 ms | 200–400 ms (plateau) |
| Plateau mechanism | — | L-type Ca²⁺ channels open → prolonged depolarization |
| Absolute refractory period | ~1 ms | ~250 ms (equal to systole — prevents tetany) |
The long plateau in cardiac AP is caused by:
- Prolonged Ca²⁺ influx through slow voltage-gated L-type Ca²⁺ channels (depolarizing)
- Simultaneous K⁺ channels are initially suppressed (preventing immediate repolarization)
- Eventually delayed rectifier K⁺ channels open → repolarization
Why does cardiac muscle need a long refractory period? It prevents summation/tetany — the heart MUST relax between contractions to refill with blood.
Smooth Muscle
- Resting potential: −50 to −60 mV (less negative than skeletal/cardiac)
- Some smooth muscle generates spontaneous pacemaker potentials (unstable resting potential)
- Action potentials may use Ca²⁺ as the primary depolarizing ion (not Na⁺)
- Multi-unit smooth muscle may depolarize without action potentials (graded potentials only)
SECTION 13: GRADED POTENTIALS vs. ACTION POTENTIALS
| Feature | Graded Potential | Action Potential |
|---|---|---|
| Amplitude | Proportional to stimulus strength | All-or-none (fixed size) |
| Propagation | Decremental (fades with distance) | Self-regenerating, non-decremental |
| Duration | Variable | Fixed (~1 ms in neurons) |
| Summation | Can summate (temporal + spatial) | Cannot summate |
| Location | Dendrites, soma, receptor endings | Axon hillock → axon |
| Purpose | Signal integration | Signal transmission over long distances |
Graded potentials summate at the axon hillock — if the summed depolarization reaches threshold (−55 mV), an action potential fires.
SECTION 14: CLINICAL AND PHARMACOLOGICAL RELEVANCE
| Drug/Toxin | Mechanism | Effect on Membrane Potential |
|---|---|---|
| Local anesthetics (lidocaine, bupivacaine) | Block voltage-gated Na⁺ channels | Prevent action potential generation → analgesia |
| Tetrodotoxin (TTX) (puffer fish toxin) | Blocks Na⁺ channel pore (outer) | Complete block of action potentials → paralysis |
| Saxitoxin (shellfish poisoning) | Same as TTX | Same effect |
| Tetraethylammonium (TEA) | Blocks K⁺ channels | Prolonged action potential |
| Digitalis | Inhibits Na⁺-K⁺ ATPase pump | ↑ intracellular Na⁺ → ↑ Ca²⁺ → ↑ cardiac contractility |
| Hyperkalemia | ↑ extracellular K⁺ → reduced K⁺ gradient → depolarizes resting potential toward threshold | Arrhythmias, cardiac arrest |
| Hypokalemia | ↓ extracellular K⁺ → hyperpolarizes membrane | Muscle weakness, cardiac arrhythmias |
| Hypocalcemia | Ca²⁺ normally stabilizes Na⁺ channel gates; ↓ Ca²⁺ → channels open at more negative potential | ↑ excitability → tetany |
SECTION 15: SUMMARY — KEY VALUES TO MEMORIZE
| Parameter | Value |
|---|---|
| Resting membrane potential (large nerve) | −70 mV |
| Threshold potential | −55 mV |
| Peak action potential overshoot | +35 mV |
| Na⁺ Nernst potential | +61 mV |
| K⁺ Nernst potential | −94 mV |
| Na⁺ pump ratio | 3 Na⁺ out : 2 K⁺ in per ATP |
| Duration of nerve action potential | ~1 ms |
| Duration of cardiac action potential | 200–400 ms |
| Absolute refractory period (nerve) | ~1 ms |
| Max firing frequency (large nerve) | ~1000 impulses/sec |
| Resting K⁺ : Na⁺ permeability ratio | ~100:1 |
| At action potential peak Na⁺ conductance increase | ~5000-fold |
SECTION 16: MEMORY AIDS
| Concept | Memory Trick |
|---|---|
| 3 Na out, 2 K in | "NaKed truth — Na leaves 3, K enters 2" |
| K⁺ determines resting potential | K⁺ Nernst = −94 mV; resting = −70 mV; close because membrane is mostly K⁺-permeable at rest |
| Na⁺ determines peak of AP | Na⁺ Nernst = +61 mV; overshoot = +35 mV; close because Na⁺ permeability skyrockets during depolarization |
| Threshold = −55 mV | "−55 is the firing line — cross it and there's no going back" |
| Inactivation gate = refractory period | "The Na⁺ channel is like a door that slams shut and needs a key (repolarization) to reset" |
| Saltatory conduction | "Jumps from node to node — faster, cheaper, cleaner" |
| Hypocalcemia → tetany | "Ca²⁺ stabilizes channels; remove it → gates open too easily → muscle spasms" |
Source: Guyton and Hall Textbook of Medical Physiology (14th Ed.), Chapter 5 (Membrane Potentials and Action Potentials) and Chapter 6 (Contraction of Skeletal Muscle).
This is a shared conversation. Sign in to Orris to start your own chat.