Pathophysiology of asthma
Reading File
Finding Sources
Finding Sources
Searching PubMed
"asthma"[MeSH Terms] AND "pathophysiology"[MeSH Terms]
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
I now have everything needed for a thorough, well-illustrated answer. Here is the full pathophysiology of asthma:
Pathophysiology of Asthma
Asthma is a chronic inflammatory airway disease characterized by variable, reversible airflow obstruction and bronchial hyperresponsiveness. Unlike COPD, it is generally not progressive - but if untreated, it leads to permanent structural change (airway remodeling).
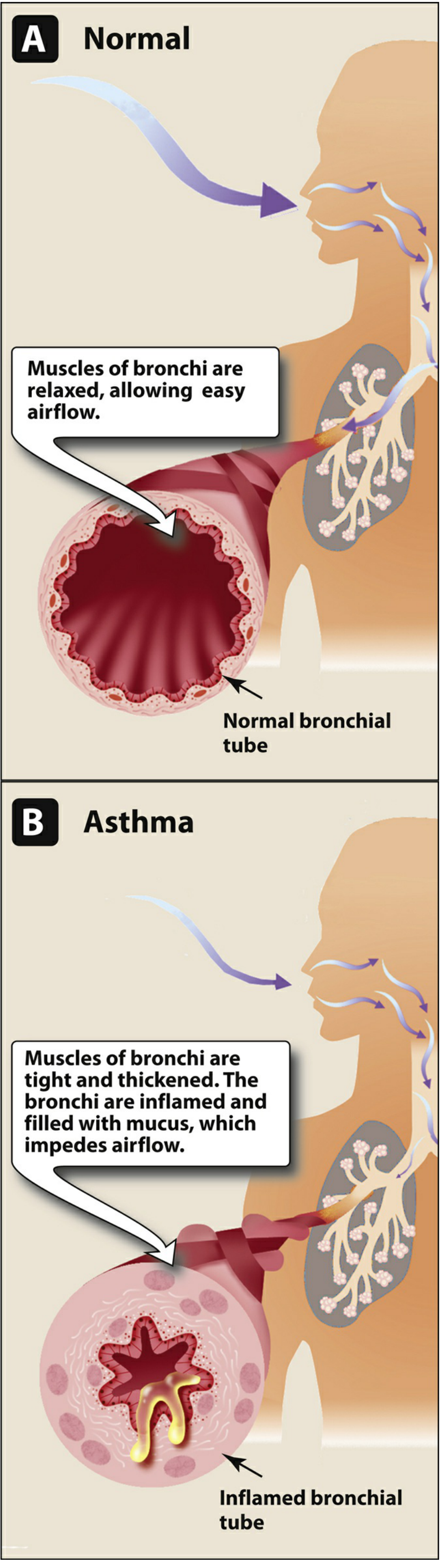
1. Overview of Airflow Obstruction
The pathophysiologic hallmark is reduction in airway diameter caused by four concurrent processes (Tintinalli's EM):
| Mechanism | Description |
|---|---|
| Smooth muscle contraction | Bronchospasm - the dominant, reversible component |
| Mucosal edema | Vascular congestion and wall swelling |
| Mucus hypersecretion | Plugs composed of mucus, serum proteins, inflammatory cells, and cellular debris |
| Inflammatory cell infiltration | Eosinophils, lymphocytes, mast cells, macrophages, dendritic cells, myofibroblasts |
These changes produce increased work of breathing, abnormal pulmonary blood flow distribution, and classic pulmonary function impairment (reduced FEV1, reduced FEV1/FVC ratio, air trapping). - Tintinalli's Emergency Medicine, p. 1345
2. Types of Asthma (Endotypes)
Atopic (Allergic) Asthma - Most Common
Begins in childhood, associated with IgE sensitization to environmental allergens (dust mites, pollen, animal dander, food, mold). A positive family history of atopy, allergic rhinitis, urticaria, or eczema is common.
Nonatopic (Intrinsic) Asthma
Develops in adults, no IgE sensitization (skin tests usually negative). Triggered by viral infections (especially rhinovirus) and airborne irritants (sulfur dioxide, ozone). Associated with chronic rhinosinusitis and nasal polyposis. Less responsive to inhaled glucocorticoids. Driven by innate immune mechanisms via Type 2 Innate Lymphoid Cells (ILC2). - Goldman-Cecil Medicine
Drug-Induced Asthma
Aspirin/NSAIDs (via arachidonic acid shunting toward leukotrienes), beta-blockers.
Neutrophilic/Mixed/Pauci-granulocytic Variants
Less common patterns seen in severe or occupational asthma. - Robbins Basic Pathology
3. Immune Mechanism: The Th2 Pathway
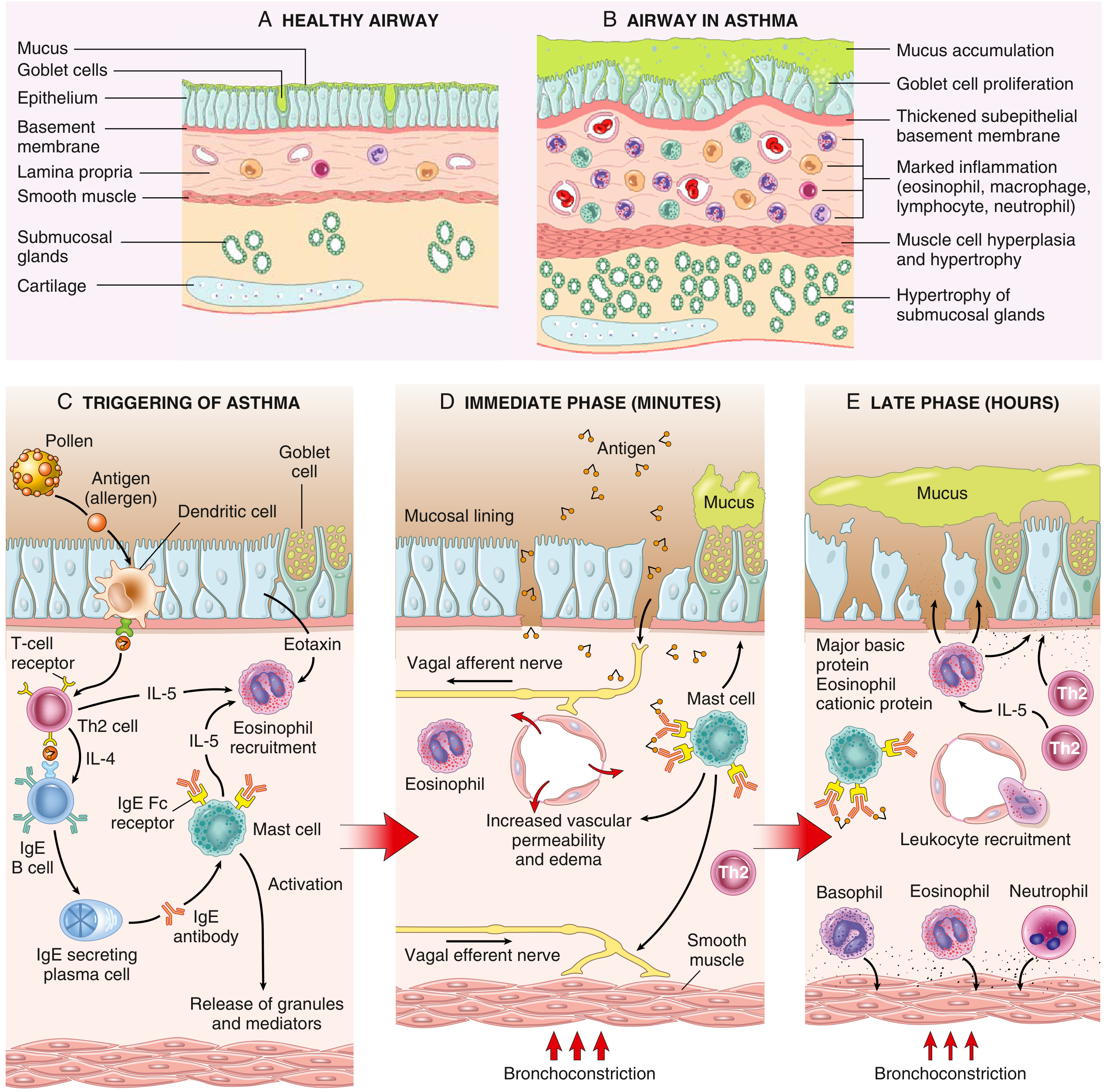
Sensitization Phase
Inhaled antigens are processed by dendritic cells, which present them to naive CD4+ T cells, skewing differentiation toward Th2 cells. Th2 cells then produce:
- IL-4 & IL-13 - Drive B cells to produce IgE; IL-13 also independently induces airway hyperresponsiveness and goblet cell metaplasia
- IL-5 - Drives eosinophil formation, maturation, recruitment, and survival
- Eotaxin - Potent chemoattractant for eosinophils
Secreted IgE binds to high-affinity Fc-epsilon receptors (FcεRI) on submucosal mast cells, "sensitizing" them. - Robbins Basic Pathology, p. 398; Goldman-Cecil Medicine
4. The Two-Wave Inflammatory Response
Early (Immediate) Phase - Within Minutes
On re-exposure to antigen, IgE molecules on mast cell surfaces are cross-linked. Mast cell degranulation releases preformed and newly synthesized mediators:
| Mediator | Effect |
|---|---|
| Histamine | Bronchoconstriction, vasodilation, increased vascular permeability |
| Leukotrienes C4, D4, E4 (cysteinyl LTs) | Potent, prolonged bronchoconstriction; mucus production |
| Prostaglandin D2 | Bronchoconstriction |
| Tryptase | Proteolytic damage; promotes airway hyperresponsiveness |
| TNF-α | Pro-inflammatory amplifier |
Bronchospasm is also reinforced by reflex vagal pathways - afferent stimulation of vagal receptors triggers efferent cholinergic bronchoconstriction. - Robbins Basic Pathology, p. 400
Late Phase - 4 to 8 Hours Later
Inflammatory mediators from the early phase stimulate epithelial cells to produce chemokines (including eotaxin), recruiting:
- Eosinophils (dominant)
- More Th2 cells
- Basophils, neutrophils, monocytes
Recruited eosinophils release major basic protein (MBP) and eosinophil cationic protein (ECP), which damage the airway epithelium, expose subepithelial nerve endings, and amplify hyperresponsiveness. This late phase sustains and amplifies inflammation beyond the initial bronchospasm. - Robbins Basic Pathology, p. 402
5. Key Cellular Players
Mast Cells
- In mild asthma: MCt type (tryptase only) in the subepithelium
- In severe asthma: MCtc type (tryptase + chymase + carboxypeptidase) increases; mast cells infiltrate airway smooth muscle bundles, directly contributing to bronchoconstriction
- Activated by IgE cross-linking but also by direct physical stimuli (cold air, exercise, osmotic changes)
Eosinophils
- Key effector cells; elevated blood eosinophils are now a biomarker predicting response to inhaled corticosteroids and anti-Th2 biologics
- Release MBP, ECP, leukotrienes, and platelet-activating factor (PAF)
- Charcot-Leyden crystals (galectin-10 from eosinophils) found in airway mucus serve as a pro-inflammatory factor
T Lymphocytes (Th2)
- Orchestrate the entire allergic cascade via IL-4, IL-5, and IL-13
Innate Lymphoid Cells Type 2 (ILC2)
- In nonatopic asthma, ILC2 cells stimulated by epithelial "alarmins" (IL-33, TSLP, IL-25) produce IL-5 and IL-13 independent of adaptive immunity - explain why up to 50% of severe asthma patients have T2 inflammation without classical IgE sensitization
Bronchial Epithelium
- Not merely a passive barrier - produces pro-inflammatory mediators after injury
- Releases alarmins (IL-33, TSLP) that activate ILC2
- Abnormal repair drives structural changes
- Fishman's Pulmonary Diseases, p. 1136-1137
6. Airway Remodeling - The Chronic Structural Consequence
Repeated cycles of inflammation lead to permanent structural changes collectively called airway remodeling. These begin early (even in childhood) and may precede clinical symptoms:
| Change | Description |
|---|---|
| Subepithelial fibrosis | Thickening of lamina reticularis from collagen types III and IV deposition |
| Smooth muscle hypertrophy & hyperplasia | Increases bronchospasm potential |
| Goblet cell hyperplasia | Mucus hypersecretion |
| Submucosal gland hypertrophy | Further mucus burden |
| Angiogenesis | New blood vessel formation in airway wall |
| Airway wall thickening | From edema and cellular infiltration; reduces lumen diameter permanently |
Remodeling can become a trigger for non-reversible loss of lung function, increasing asthma severity and mortality. - Washington Manual, p. 5161-5167; Rosen's Emergency Medicine, p. 1683
7. Triggers of Acute Exacerbation
| Category | Examples |
|---|---|
| Allergens | House dust mites, cockroaches, mold, pet dander, pollen |
| Infections | Rhinovirus (accounts for 40-80% of adult and 80% of pediatric exacerbations); rhinovirus triggers a Th1/neutrophilic response with IL-33 → ILC2 → eosinophil recruitment |
| Irritants | Tobacco smoke, ozone, NO2, perfumes, cold air |
| Exercise | Evaporative cooling/hyperosmolarity of airway lining fluid |
| Occupational | Metal salts, wood dust, isocyanates, biological enzymes |
| Drugs | Aspirin/NSAIDs, beta-blockers (including eye drops) |
| Stress | Emotional stress; endocrine factors (estradiol/progesterone fluctuations) |
| GERD | Micro-aspiration and vagal reflex |
8. Genetics
Asthma has ~60% heritability (twin studies). Genome-wide association studies (GWAS) have identified multiple loci, notably:
- Chromosome 17q21 - first robust asthma susceptibility locus
- Genes encoding the IL-4 receptor, ORMDL3/GSDMB (17q21)
- Heritability for bronchial hyperresponsiveness: 30-66%
Environmental-gene interactions (allergen load, pollution, hygiene hypothesis - microbial exposure in early life suppressing atopic expression) shape disease phenotype. - Rosen's Emergency Medicine, p. 1687
9. Pulmonary Function Consequences
- Reduced FEV1 and FEV1/FVC ratio (obstructive pattern)
- Air trapping - increased residual volume
- Obstruction is worst on expiration (dynamic airway collapse), making expiration effortful and fatiguing
- Hyperinflation from incomplete emptying
- Severe obstruction = silent chest (absent wheeze due to minimal airflow)
- Reversibility with bronchodilators is the diagnostic hallmark
Summary Diagram of Mechanism
Allergen exposure
↓
Dendritic cell → Th2 differentiation
↓
IL-4/IL-13 → IgE production by B cells
↓
IgE binds Fc receptors on MAST CELLS
↓
Re-exposure → IgE cross-linking → Degranulation
↓
EARLY PHASE (minutes): Histamine, LTC4/D4/E4, PGD2
→ Bronchoconstriction + vasodilation + mucus
↓
LATE PHASE (4-8 hrs): Chemokines, Eotaxin
→ Eosinophil, Th2 cell, basophil recruitment
→ MBP/ECP → Epithelial damage + hyperresponsiveness
↓
CHRONIC: Airway remodeling (fibrosis, hypertrophy, angiogenesis)
Sources:
- Robbins & Kumar Basic Pathology, pp. 387-428
- Goldman-Cecil Medicine, pp. 2151-2166
- Washington Manual of Medical Therapeutics, pp. 5147-5198
- Tintinalli's Emergency Medicine, pp. 1343-1354
- Rosen's Emergency Medicine, pp. 1677-1688
- Fishman's Pulmonary Diseases and Disorders, pp. 1126-1136
- Lippincott Illustrated Reviews: Pharmacology, p. 1389
This is a shared conversation. Sign in to Orris to start your own chat.