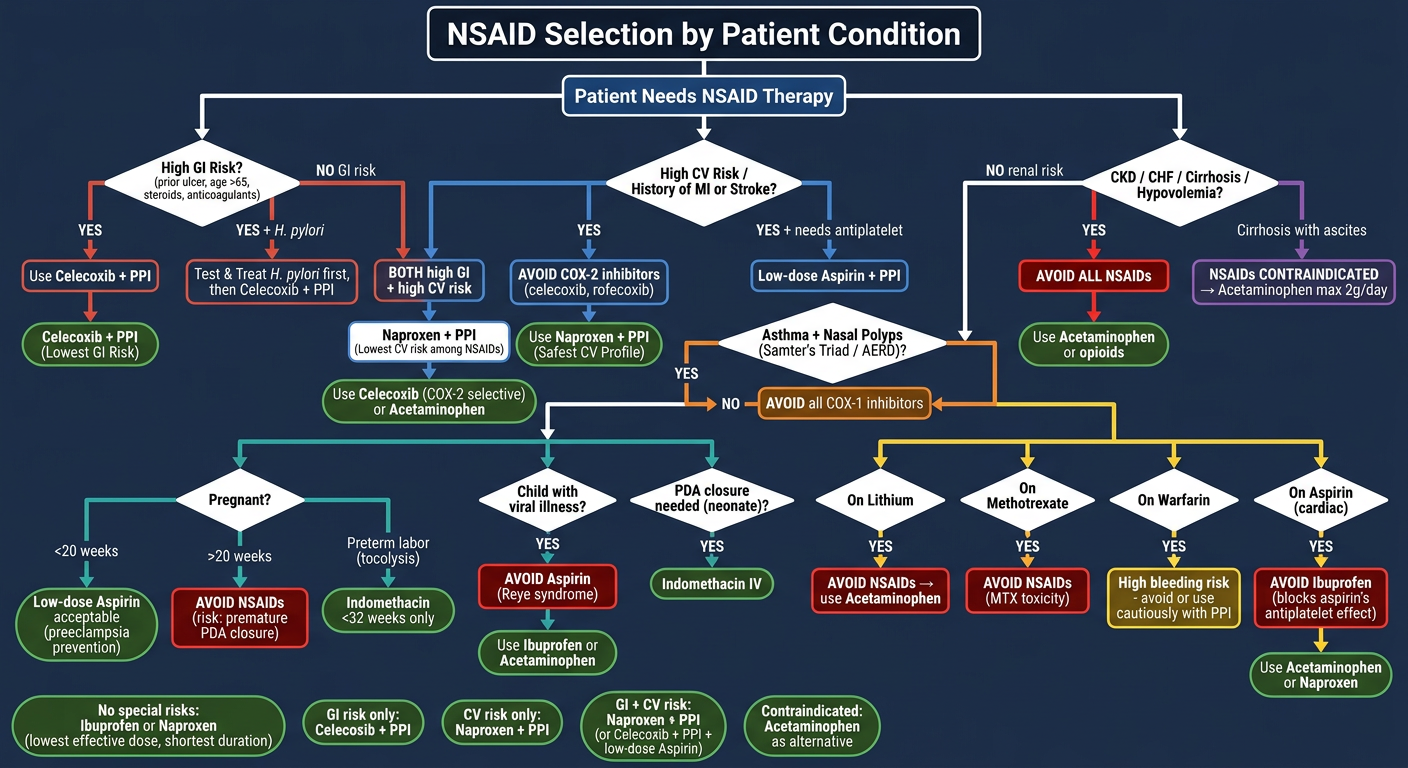
Here are USMLE-style high-yield questions on NSAIDs, organized by system:
NSAIDs - USMLE Standard Questions
MECHANISM OF ACTION
Question 1
A 55-year-old woman with rheumatoid arthritis has been taking ibuprofen for 3 years. She is scheduled for an elective knee replacement in 1 week. Her surgeon asks her to stop ibuprofen but notes she can continue her low-dose aspirin for cardiac protection. Which of the following best explains why aspirin's antiplatelet effect persists longer than ibuprofen's?
- A) Aspirin has a longer half-life than ibuprofen
- B) Aspirin irreversibly acetylates COX-1 in platelets
- C) Aspirin is more potent at COX-2 inhibition
- D) Ibuprofen is rapidly cleared by renal excretion
- E) Aspirin activates thromboxane synthesis
Answer & Explanation
Answer: B
Aspirin irreversibly acetylates the serine residue on COX-1 (and COX-2), permanently inactivating it. Since platelets are anucleate and cannot synthesize new COX, the effect lasts the entire platelet lifespan (7-10 days). All other NSAIDs (including ibuprofen) are reversible, competitive COX inhibitors, so platelet function returns as the drug clears. Half-life of aspirin (~15-20 min for the acetylsalicylate form) is actually shorter than ibuprofen (~2 hours), making choice A wrong.
Question 2
A 62-year-old man with a history of MI on daily aspirin 81 mg starts taking ibuprofen 400 mg three times daily for osteoarthritis pain. Three months later, he has a second MI. Which of the following best explains this outcome?
- A) Ibuprofen directly promotes platelet aggregation
- B) Ibuprofen irreversibly inhibits COX-2 in vascular endothelium
- C) Ibuprofen competitively blocks COX-1 and prevents aspirin from acetylating it
- D) Ibuprofen increases thromboxane A2 production
- E) Aspirin's half-life is shortened by ibuprofen
Answer & Explanation
Answer: C
This is one of the most tested NSAID drug interactions. Ibuprofen (and other NSAIDs) competitively and reversibly bind to COX-1 at the same site aspirin must acetylate. If ibuprofen is taken before aspirin, it physically occupies the serine acetylation site, preventing aspirin's irreversible binding. When ibuprofen later dissociates, the window for aspirin's cardioprotective irreversible acetylation is lost. To avoid this, aspirin should be taken at least 30-60 minutes before ibuprofen.
GI SYSTEM
Question 3
A 68-year-old man with osteoarthritis and a past history of duodenal ulcer takes diclofenac daily for knee pain. He presents to the ED with hematemesis and is found to have an actively bleeding gastric ulcer on endoscopy. Which of the following pathophysiologic mechanisms is most responsible?
- A) Direct alkaline injury to gastric mucosa
- B) Inhibition of COX-2 in the gastric lining
- C) Decreased mucosal PGE2 → reduced mucus/bicarbonate secretion
- D) Increased gastrin secretion
- E) Stimulation of parietal cell H+/K+ ATPase
Answer & Explanation
Answer: C
PGE2 (and PGI2), synthesized via COX-1 in gastric mucosa, maintain the mucosal defense barrier by stimulating mucus and bicarbonate secretion, promoting mucosal blood flow, and facilitating epithelial repair. NSAIDs inhibit COX-1, depleting these prostaglandins. This is the primary systemic mechanism of NSAID-induced ulceration. Note: NSAIDs also cause topical injury (being weak acids that penetrate epithelial cells), but the dominant and most tested mechanism is the prostaglandin-depleting systemic effect.
Question 4
A 70-year-old woman with rheumatoid arthritis and a history of peptic ulcer disease requires long-term NSAID therapy. Which of the following strategies is MOST appropriate to reduce her GI risk?
- A) Prescribe indomethacin with antacids
- B) Prescribe celecoxib with a proton pump inhibitor
- C) Prescribe naproxen alone as it has the lowest GI toxicity
- D) Prescribe ibuprofen at the maximum dose for best efficacy
- E) Add H2 blocker to any non-selective NSAID
Answer & Explanation
Answer: B
In patients at high GI risk (prior ulcer, age >65, concurrent anticoagulant or steroid use), the preferred strategy is a selective COX-2 inhibitor (celecoxib) plus a PPI. Celecoxib spares COX-1-mediated gastric protection, and the PPI provides additional acid suppression. Misoprostol (PGE1 analog) is an alternative to PPI. Naproxen has a relatively better cardiovascular profile but is NOT the GI-safest NSAID - celecoxib is. Indomethacin has the highest GI toxicity.
Question 5
A 59-year-old man on daily aspirin and an SSRI presents with melena. Upper endoscopy reveals a bleeding gastric ulcer. His physician notes the SSRI increased his bleeding risk. Which mechanism explains the SSRI's contribution?
- A) SSRIs inhibit platelet COX-1
- B) SSRIs deplete platelet serotonin stores, impairing platelet aggregation
- C) SSRIs induce CYP enzymes that activate aspirin
- D) SSRIs decrease gastrin production
- E) SSRIs cause direct gastric mucosal erosion
Answer & Explanation
Answer: B
Platelets take up serotonin via SERT (the same transporter SSRIs block). Platelet serotonin is released during activation and amplifies aggregation. SSRIs deplete platelet serotonin, impairing this step. This effect is synergistic with NSAIDs' antiplatelet and mucosal effects, and the combination significantly increases GI bleeding risk.
CARDIOVASCULAR SYSTEM
Question 6
A 64-year-old woman with osteoarthritis, controlled hypertension on lisinopril, and a history of GERD is started on celecoxib for joint pain. Two months later, her blood pressure is 158/96 mmHg. Which of the following best explains this finding?
- A) Celecoxib activates the renin-angiotensin system directly
- B) COX-2 inhibition reduces renal PGI2 → sodium retention → hypertension
- C) Celecoxib inhibits lisinopril absorption
- D) Celecoxib increases aldosterone secretion
- E) COX-2 inhibition increases thromboxane A2 in the vasculature
Answer & Explanation
Answer: B
COX-2 is constitutively expressed in the kidney. Inhibiting renal COX-2 reduces vasodilatory prostaglandins (PGE2, PGI2), leading to sodium and water retention, reduced efficacy of antihypertensives, and elevated blood pressure. This is a class effect of ALL NSAIDs, not just selective COX-2 inhibitors. NSAIDs blunt the effect of ACE inhibitors, ARBs, diuretics, and beta-blockers.
Question 7
A pharmaceutical company withdraws a COX-2 selective inhibitor from the market after post-marketing surveillance shows increased rates of myocardial infarction. Which of the following best explains the cardiovascular risk of selective COX-2 inhibitors?
- A) Selective inhibition of thromboxane A2 production
- B) Inhibition of PGI2 (prostacyclin) without inhibiting platelet TxA2
- C) Increased platelet adhesion via COX-2 upregulation
- D) Direct vasoconstriction via COX-2 in smooth muscle
- E) Increased fibrinogen synthesis
Answer & Explanation
Answer: B
The balance between PGI2 (prostacyclin) and TxA2 is the key concept. PGI2 is synthesized via COX-2 in vascular endothelium - it is vasodilatory and anti-aggregatory. TxA2 is synthesized via COX-1 in platelets - it is vasoconstricting and pro-aggregatory. Selective COX-2 inhibitors suppress PGI2 but leave platelet TxA2 (COX-1) intact, tipping the balance toward thrombosis, vasoconstriction, and ultimately MI/stroke. This is exactly what happened with rofecoxib (Vioxx).
RENAL SYSTEM
Question 8
A 72-year-old man with compensated systolic heart failure (EF 30%) and mild CKD (creatinine 1.6 mg/dL) is started on ibuprofen 400 mg TID by his PCP for back pain. One week later he presents with 3+ pitting edema, decreased urine output, and creatinine of 3.8 mg/dL. Which of the following best explains this deterioration?
- A) Ibuprofen directly causes tubular necrosis
- B) Ibuprofen inhibits renal prostaglandins that were maintaining GFR via afferent arteriolar vasodilation
- C) Ibuprofen blocks ENaC channels in the collecting duct
- D) Ibuprofen activates the macula densa to reduce GFR
- E) Ibuprofen-induced hepatotoxicity reduces oncotic pressure
Answer & Explanation
Answer: B
In patients with reduced cardiac output (CHF), hypovolemia, cirrhosis, or CKD, the kidney relies on prostaglandin-mediated vasodilation of the afferent arteriole to maintain adequate GFR despite elevated angiotensin II and sympathetic tone. NSAIDs inhibit this compensatory vasodilation, causing hemodynamically mediated pre-renal AKI. In a normal healthy kidney, prostaglandins play a minor role and NSAIDs rarely cause AKI - this distinction is a classic USMLE concept.
Question 9
A 66-year-old man with CKD stage 3 and type 2 diabetes is started on naproxen. Two weeks later, labs show potassium of 6.2 mEq/L (previously 4.8 mEq/L). Which mechanism best explains his hyperkalemia?
- A) Direct potassium release from muscle cells
- B) ↓ Renal prostaglandins → ↓ renin → ↓ aldosterone → reduced potassium excretion
- C) Naproxen blocks K+/H+ ATPase in the distal nephron
- D) NSAIDs directly bind to aldosterone receptors
- E) Hemolysis from NSAID-induced RBC damage
Answer & Explanation
Answer: B
NSAIDs inhibit renal prostaglandins, which normally stimulate renin secretion from juxtaglomerular cells. Reduced renin → reduced angiotensin II → reduced aldosterone → reduced K+ excretion in collecting duct → hyperkalemia. This is a type IV renal tubular acidosis (hyporeninemic hypoaldosteronism) pattern. Risk is highest in diabetics and CKD patients already prone to low renin states. Adding an ACE inhibitor or ARB to NSAIDs dramatically worsens this.
HEMATOLOGIC SYSTEM
Question 10
A 45-year-old woman is scheduled for a laparoscopic cholecystectomy in 5 days. She takes aspirin 81 mg daily for secondary prevention of MI (she had an MI 2 years ago). Her surgeon asks whether to hold aspirin. What is the correct recommendation?
- A) Hold aspirin 5 days before surgery to reduce bleeding risk
- B) Continue aspirin perioperatively; the cardiovascular risk of stopping outweighs surgical bleeding risk
- C) Switch to clopidogrel preoperatively
- D) Hold aspirin and give fresh frozen plasma before surgery
- E) Double the aspirin dose to ensure adequate platelet recovery
Answer & Explanation
Answer: B
For patients on aspirin for secondary prevention (established CAD, prior MI), current guidelines generally recommend continuing aspirin perioperatively for most low-to-intermediate bleeding-risk surgeries. The risk of an acute coronary event from stopping antiplatelet therapy exceeds the marginal increase in surgical bleeding. Holding aspirin is appropriate in high-bleeding-risk procedures (e.g., neurosurgery, intracranial). This is a high-yield USMLE management question distinguishing primary vs. secondary prevention.
RESPIRATORY SYSTEM
Question 11
A 38-year-old woman presents with recurrent episodes of severe bronchospasm, nasal congestion, and rhinorrhea triggered each time she takes ibuprofen for headaches. She also has nasal polyps on exam. Which of the following is the most likely pathophysiologic mechanism?
- A) IgE-mediated mast cell degranulation to ibuprofen
- B) COX-1 inhibition diverts arachidonic acid to lipoxygenase pathway → excess leukotrienes → bronchoconstriction
- C) COX-2 inhibition decreases bronchodilatory prostaglandins
- D) Direct histamine release from ibuprofen
- E) Ibuprofen-induced airway eosinophilic granulomatosis
Answer & Explanation
Answer: B
This is Samter's Triad (Aspirin-Exacerbated Respiratory Disease, AERD): asthma + nasal polyps + NSAID sensitivity. The mechanism is NOT immunologic/IgE-mediated (not a true allergy). COX-1 inhibition blocks the prostaglandin pathway, shunting arachidonic acid to the 5-lipoxygenase pathway → overproduction of cysteinyl leukotrienes (LTC4, LTD4, LTE4) → potent bronchoconstriction and mucus secretion. Safe alternatives include celecoxib (COX-2 selective) and acetaminophen (at standard doses).
Question 12
A 10-year-old boy is brought to the ED with altered mental status, vomiting, and jaundice that developed 5 days after his parents gave him aspirin for fever associated with a flu-like illness. Labs show elevated ammonia, elevated LFTs, hypoglycemia, and prolonged PT. Which of the following best describes this condition?
- A) Aspirin overdose causing salicylate toxicity
- B) Reye's syndrome - mitochondrial dysfunction from aspirin use during viral illness
- C) Fulminant hepatitis A from the viral infection
- D) Acetaminophen hepatotoxicity from overdose
- E) Autoimmune hepatitis triggered by viral illness
Answer & Explanation
Answer: B
Reye's syndrome is characterized by acute non-inflammatory encephalopathy and fatty degeneration of the liver following aspirin use in children during viral illnesses (especially influenza B and varicella). Key features: elevated ammonia, liver dysfunction, hypoglycemia, normal bilirubin (non-icteric usually), microvesicular fatty change on liver biopsy. Aspirin is contraindicated in children under 18 years with febrile viral illnesses for this reason. This is why acetaminophen or ibuprofen is used instead for pediatric fever.
OBSTETRICS / PREGNANCY
Question 13
A 28-year-old woman at 34 weeks gestation presents in preterm labor with regular contractions every 5 minutes. Her cervix is 2 cm dilated. The obstetrician considers tocolysis. Which NSAID is used as a tocolytic, and what is the primary fetal concern limiting its use after 32 weeks?
- A) Celecoxib; risk of neonatal pulmonary hypertension
- B) Indomethacin; premature closure of the ductus arteriosus
- C) Ibuprofen; neonatal renal aplasia
- D) Ketorolac; intracerebral hemorrhage
- E) Aspirin; placental abruption
Answer & Explanation
Answer: B
Indomethacin is used as a tocolytic (to delay preterm labor) because prostaglandins (PGE2, PGF2α) are required for myometrial contractions. By inhibiting prostaglandin synthesis, indomethacin reduces contractions. However, the ductus arteriosus in the fetus remains patent via PGE2. Indomethacin can cause premature ductal closure → fetal pulmonary hypertension. This risk increases significantly after 32 weeks' gestation, limiting its use. Oligohydramnios (via reduced fetal urine output from renal prostaglandin inhibition) is another concern.
Question 14
A 31-year-old woman at 26 weeks gestation with a history of two prior pregnancy losses is started on low-dose aspirin. Her obstetrician explains this reduces her risk for a dangerous complication. What is the indication, and what is the mechanism?
- A) Prevention of gestational diabetes; aspirin improves insulin sensitivity
- B) Prevention of preeclampsia; low-dose aspirin inhibits TxA2-mediated platelet aggregation and vasoconstriction
- C) Prevention of placental abruption; aspirin promotes fibrinolysis
- D) Prevention of preterm labor; aspirin inhibits uterine contractions
- E) Prevention of IUGR; aspirin increases placental blood volume
Answer & Explanation
Answer: B
Low-dose aspirin (81 mg/day) is recommended for prevention of preeclampsia in high-risk women (prior preeclampsia, chronic hypertension, multifetal gestation, renal disease, etc.) starting at 12-16 weeks. Preeclampsia involves an imbalance between TxA2 (pro-aggregatory, vasoconstrictive - COX-1 in platelets) and PGI2 (vasodilatory, anti-aggregatory). Low-dose aspirin preferentially inhibits platelet COX-1 → ↓ TxA2 → restores the balance. The dose is low enough to spare endothelial PGI2 production (which requires higher COX activity for resynthesis).
CNS / TOXICOLOGY
Question 15
A 22-year-old woman intentionally ingests a large amount of aspirin. In the ED, she is tachypneic with a respiratory rate of 32, confused, and diaphoretic. ABG shows pH 7.49, PaCO2 28, PaO2 98, HCO3 21. Two hours later, repeat ABG shows pH 7.28, PaCO2 34, HCO3 15. What is the correct sequence of acid-base disturbances?
- A) Metabolic acidosis → respiratory alkalosis
- B) Respiratory alkalosis → mixed metabolic acidosis + respiratory alkalosis
- C) Metabolic alkalosis → metabolic acidosis
- D) Respiratory acidosis → metabolic acidosis
- E) Normal anion gap metabolic acidosis throughout
Answer & Explanation
Answer: B
Salicylate toxicity produces a characteristic acid-base progression:
- Early: Salicylates directly stimulate the medullary respiratory center → primary respiratory alkalosis (hyperventilation, ↑ pH, ↓ PaCO2)
- Late: Salicylates uncouple oxidative phosphorylation → accumulation of organic acids (lactic acid, ketoacids) + direct organic acid load → high anion gap metabolic acidosis superimposed on the respiratory alkalosis.
In adults, the mixed picture (respiratory alkalosis + metabolic acidosis) is classic. In children, pure metabolic acidosis is more common early. Treatment: sodium bicarbonate infusion (alkalinizes urine, traps ionized salicylate in renal tubule → enhances excretion); hemodialysis in severe cases.
DRUG INTERACTIONS
Question 16
A 58-year-old man with bipolar disorder on lithium carbonate is started on ibuprofen for knee arthritis by a different provider. Two weeks later he presents with coarse tremor, confusion, and nausea. His lithium level is 2.8 mEq/L (therapeutic: 0.6-1.2). What is the mechanism of this interaction?
- A) Ibuprofen displaces lithium from plasma proteins
- B) NSAIDs inhibit renal prostaglandins → ↓ GFR and reduced lithium excretion → lithium accumulation
- C) Ibuprofen induces CYP enzymes that convert lithium to a toxic metabolite
- D) Ibuprofen directly inhibits lithium tubular secretion at the PCT
- E) NSAID-induced diarrhea increases lithium absorption
Answer & Explanation
Answer: B
Lithium is excreted renally and handled similarly to sodium. NSAIDs reduce GFR by inhibiting renal prostaglandins, decreasing lithium clearance. Additionally, any NSAID-induced sodium retention signals the proximal tubule to reabsorb more sodium (and lithium along with it), further increasing lithium levels. This is a classic and frequently tested NSAID drug interaction. NSAIDs are essentially contraindicated in patients on lithium, or require very close monitoring and dose adjustment.
Question 17
A 45-year-old woman with rheumatoid arthritis is on weekly low-dose methotrexate. Her rheumatologist adds naproxen for acute joint pain. What is the primary danger of this combination?
- A) Naproxen induces hepatic metabolism of methotrexate → sub-therapeutic levels
- B) NSAIDs reduce renal elimination of methotrexate → methotrexate toxicity (mucositis, pancytopenia)
- C) NSAIDs increase methotrexate absorption from the GI tract
- D) Naproxen competes with methotrexate for dihydrofolate reductase
- E) This is a safe combination routinely used in rheumatology
Answer & Explanation
Answer: B
NSAIDs reduce renal blood flow (by inhibiting prostaglandins) and may compete with methotrexate for tubular secretion. Both effects reduce methotrexate clearance, leading to accumulation and toxicity - manifest as mucositis, bone marrow suppression (pancytopenia), hepatotoxicity, and rarely pneumonitis. This is a dangerous combination, especially at high methotrexate doses. At low rheumatologic doses (7.5-25 mg/week), the combination is used with caution and monitoring, but the examiner will often test whether you recognize the mechanism of toxicity.
RENAL / UROLOGY
Question 18
A 26-year-old premature neonate (32 weeks gestational age) has persistent tachycardia, a wide pulse pressure, and a continuous "machinery" murmur. Echocardiography confirms a patent ductus arteriosus with left-to-right shunting. Which drug is indicated for pharmacologic closure, and what is its mechanism?
- A) Prostaglandin E1 (alprostadil); promotes ductal smooth muscle relaxation
- B) Indomethacin; inhibits COX → ↓ PGE2 → ductal smooth muscle constriction
- C) Furosemide; reduces left-to-right shunt by decreasing preload
- D) Digoxin; increases cardiac output and reduces shunt
- E) Sildenafil; inhibits PDE5 → promotes ductal closure
Answer & Explanation
Answer: B
The ductus arteriosus remains patent due to circulating PGE2 (and low oxygen tension in fetal life). Indomethacin (or ibuprofen) inhibits COX → reduces PGE2 → ductal smooth muscle contracts → PDA closes. This is the pharmacologic treatment of choice for PDA in premature neonates. Contraindications to indomethacin: renal failure, thrombocytopenia, necrotizing enterocolitis, active bleeding. Note: PGE1 (alprostadil) does the opposite - it KEEPS the ductus open (used in duct-dependent congenital heart defects like critical pulmonic stenosis or TGA to maintain ductal patency until surgery).
BONUS - RAPID FIRE
Question 19
A patient with a known sulfonamide drug allergy asks about taking celecoxib. The physician is cautious because:
- A) Celecoxib is metabolized to a sulfonamide metabolite
- B) Celecoxib contains a sulfonamide moiety (benzenesulfonamide group) that may cause cross-reactivity
- C) Celecoxib inhibits folate synthesis like sulfonamides
- D) No caution is needed; there is no structural relationship
- E) Celecoxib is contraindicated in all allergy patients
Answer & Explanation
Answer: B
Celecoxib is a diaryl-substituted pyrazole with a sulfonamide side chain (benzenesulfonamide). Patients with sulfonamide hypersensitivity reactions may theoretically cross-react. In practice the risk is low (sulfonamide antibiotics vs. non-antibiotic sulfonamides have different allergy profiles), but the USMLE expects you to know that celecoxib is structurally a sulfonamide derivative and should be used with caution in sulfa-allergic patients.
Question 20
A 55-year-old patient with liver cirrhosis and ascites asks whether he can take ibuprofen for his chronic back pain. The BEST response is:
- A) It is safe to use at half the normal dose
- B) It can be used intermittently but not daily
- C) NSAIDs are contraindicated; the kidney depends on prostaglandins for GFR in cirrhosis
- D) Use only topical NSAIDs to avoid systemic effects
- E) Switch to indomethacin which is safer in liver disease
Answer & Explanation
Answer: C
NSAIDs are contraindicated in cirrhosis with ascites. Patients with cirrhosis have splanchnic vasodilation and reduced effective circulating volume; they compensate by increasing renal prostaglandin synthesis to maintain renal perfusion. NSAIDs inhibit this compensatory mechanism → AKI, worsening ascites, hyponatremia, and hepatorenal syndrome. Acetaminophen (at reduced doses, e.g., 2g/day maximum) is the preferred analgesic in cirrhosis for mild-moderate pain.
20 questions covering all major NSAID topics tested on USMLE Step 1 and Step 2 CK. Sources: Katzung's Basic & Clinical Pharmacology 16e, Harrison's Principles 22e, Goodman & Gilman's Pharmacological Basis of Therapeutics, Robbins & Cotran Pathologic Basis of Disease.