Lupus nephritis
Reading File
Finding Sources
Searching PubMed
"lupus nephritis"[MeSH Terms] AND ("treatment"[MeSH Terms] OR "management")
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Reading File
Loading Image
Finding Sources
I have all the material needed for a comprehensive answer. Let me now compose it.
Lupus Nephritis
Lupus nephritis (LN) is renal involvement in systemic lupus erythematosus (SLE). It is the single greatest contributor to morbidity and mortality in lupus patients, affecting approximately 30% of people with SLE. It is most common in Black females aged 15-45 years.
Epidemiology & Risk Factors
- More prevalent and more severe in Asian and Black populations compared to Caucasians; in China, Korea, and Japan, LN accounts for >50% of all secondary glomerular diseases
- Higher genetic risk burden in East Asian populations (more risk variant alleles from GWAS data)
-
50 genetic polymorphisms linked to LN susceptibility, including variants in CCR6 (a Th17 surface marker)
- Disease onset after age 50 is associated with more chronic damage and inferior outcomes
- Brenner and Rector's The Kidney, p. 3380
Pathogenesis
LN involves both immune and non-immune mechanisms:
Immune mechanisms:
- Circulating autoantibodies to nuclear antigens (ANAs) - most specific are anti-dsDNA antibodies; others include anti-histone, anti-RNP, anti-Sm, anti-C1q, and anti-endothelial antibodies
- Glomerular immune complex deposition with complement activation (classical pathway via C1q)
- B- and T-lymphocyte dysregulation; macrophage and dendritic cell involvement
- Neutrophil extracellular traps (NETs) and apoptotic pathway involvement
- Infiltration of kidneys by neutrophils, T cells, B cells, macrophages, and dendritic cells
- Key mediators: chemokines, interferons (especially type I), and interferon regulatory factors
Non-immune mechanisms:
- Disruption of cell-cell interactions
- Vascular injury and hypoxia
- Glomerulosclerosis and interstitial fibrosis
Single-cell RNA-seq profiling of LN biopsies reveals multiple T-cell, B-cell, NK-cell, and myeloid populations, with prominent interferon responses and expression of chemokine receptors CXCR4 and CX3CR1.
- Henry's Clinical Diagnosis and Management, p. 1266-1267
ISN/RPS Classification (2003, revised 2018)
The International Society of Nephrology / Renal Pathology Society classification has six classes, assessed primarily by light microscopy (LM), immunofluorescence (IF), and electron microscopy (EM):
| Class | Name | Key Features |
|---|---|---|
| I | Minimal mesangial LN | Normal LM; mesangial deposits on IF and EM only |
| II | Mesangial proliferative LN | Mesangial hypercellularity on LM; mesangial deposits on IF/EM |
| III | Focal LN | Endocapillary hypercellularity in <50% of glomeruli; segmental, often with necrosis/crescents; focal mesangial and capillary wall deposits |
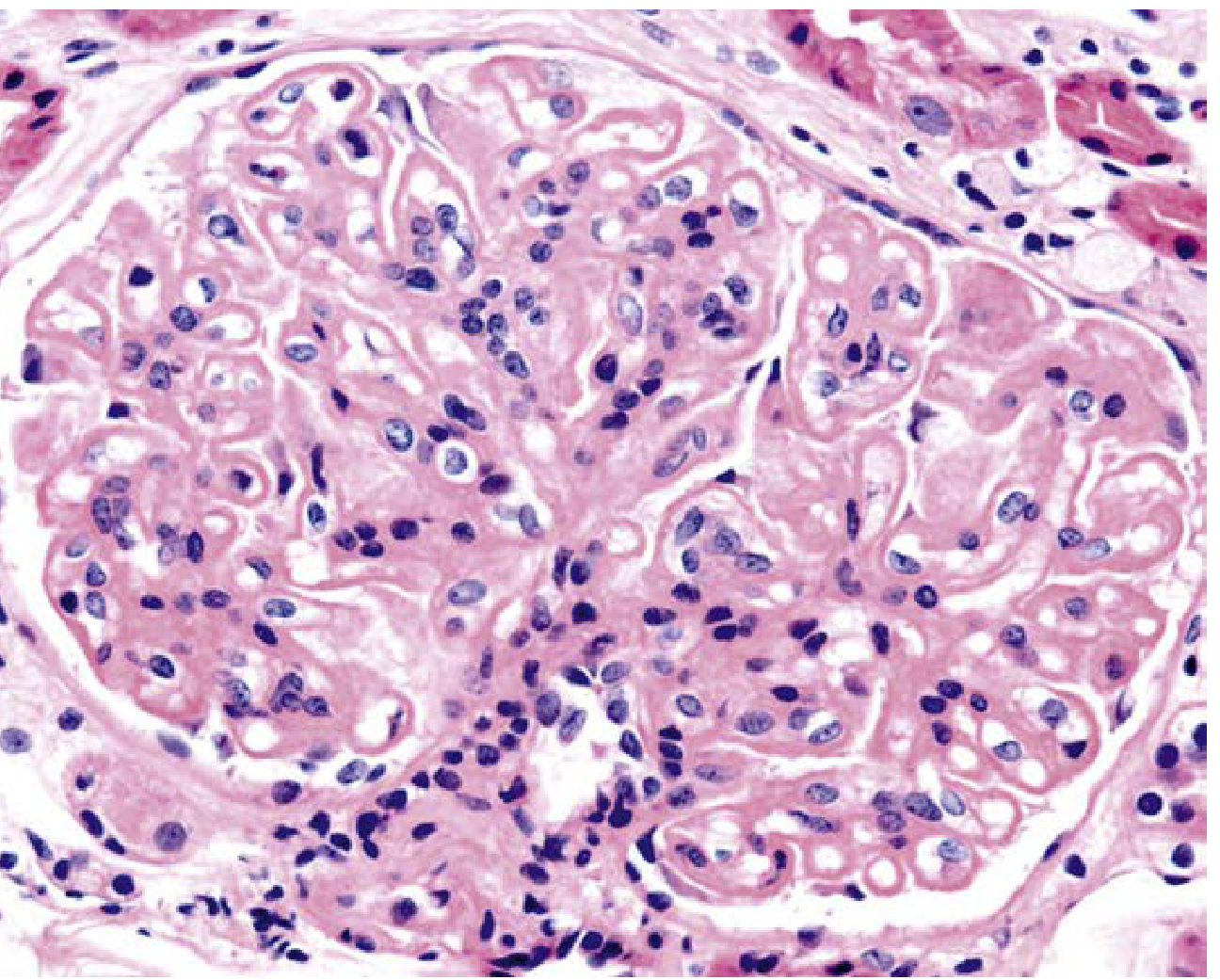
| IV | Diffuse LN | Endocapillary hypercellularity in >50% of glomeruli; "wire loops"; karyorrhectic debris; hyaline thrombi; most severe class |
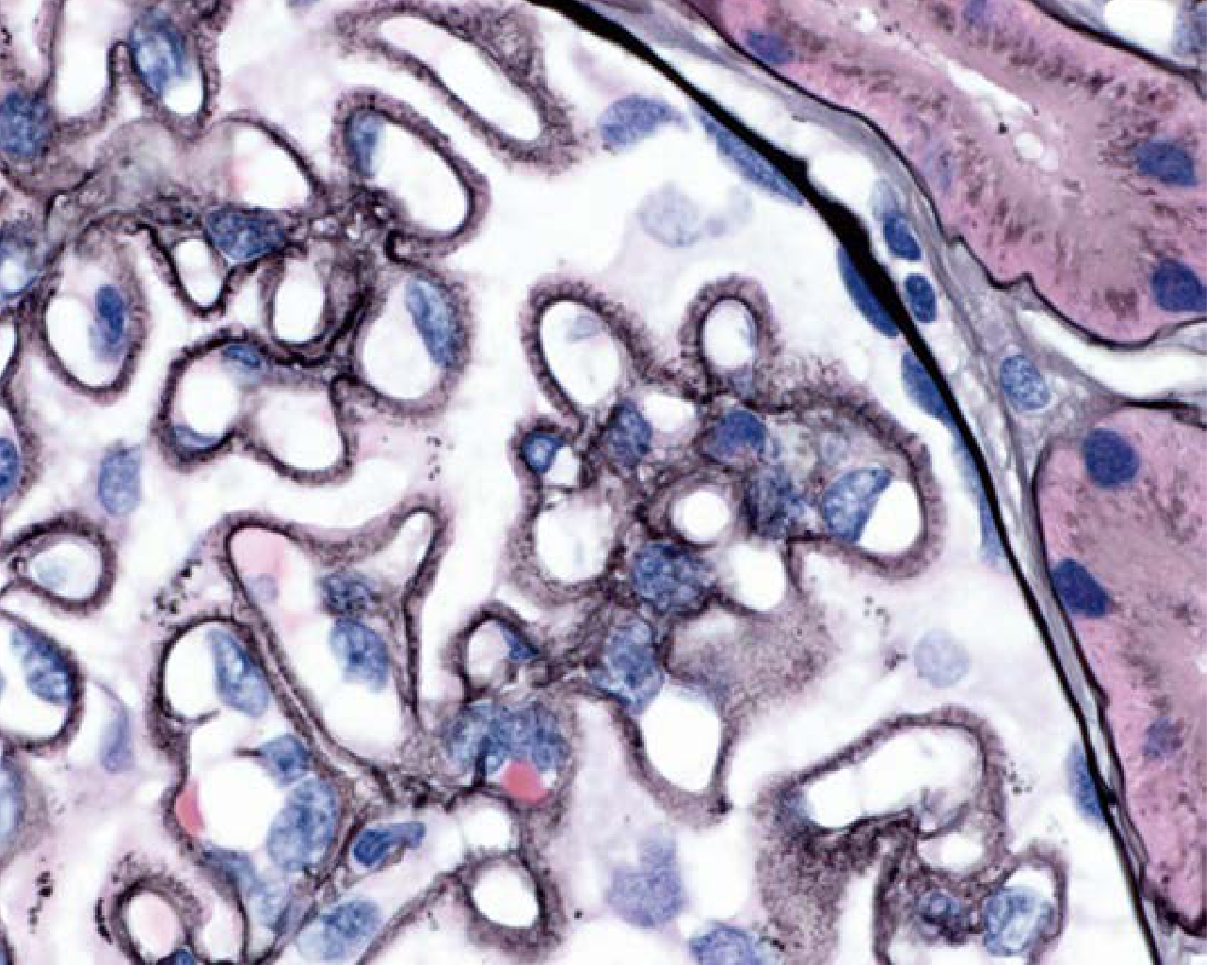
| V | Membranous LN | Thickened capillary walls; GBM vacuoles/spikes on silver stain; subepithelial deposits on EM; can occur with class III or IV |
| VI | Advanced sclerosing LN | >90% global glomerulosclerosis, no residual activity |
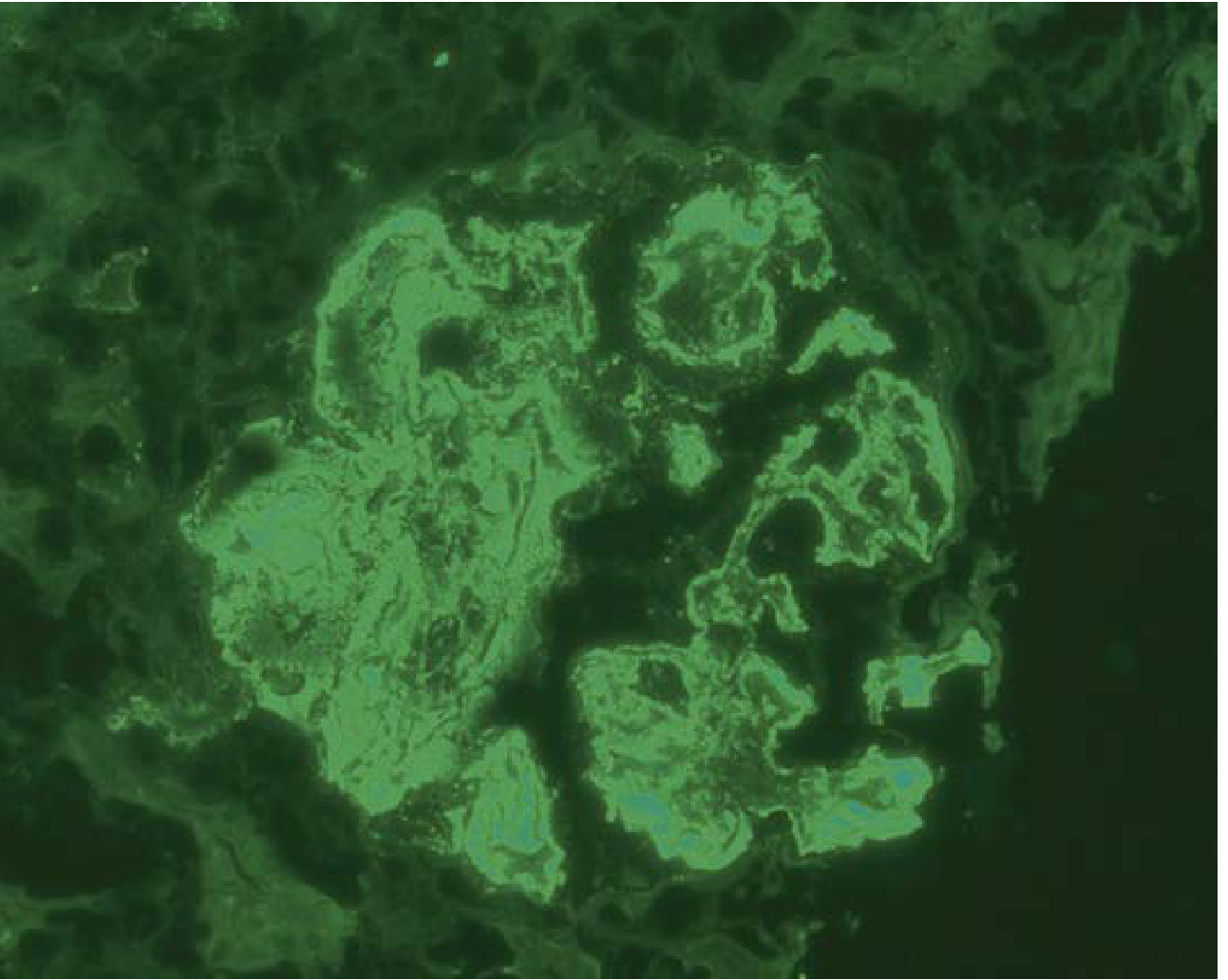
Key immunofluorescence finding: All LN classes show characteristic "full house" staining - positivity for all immunoglobulins (IgG, IgM, IgA) and complement components (C3, C1q). Intense C1q staining is a diagnostically helpful hallmark.
Classes III and IV are qualitatively similar - the distinction is purely quantitative (< vs. >50% glomerular involvement).
Lupus podocytopathy - a separate entity not in the ISN/RPS classification - presents with nephrotic syndrome, FSGS or minimal change-like lesions on LM, and severe podocyte foot process effacement on EM without subendothelial deposits. High relapse rate.
- Henry's Clinical Diagnosis and Management, pp. 1261-1267
Histological Images
Class I - IgG immunofluorescence (mesangial deposits only):
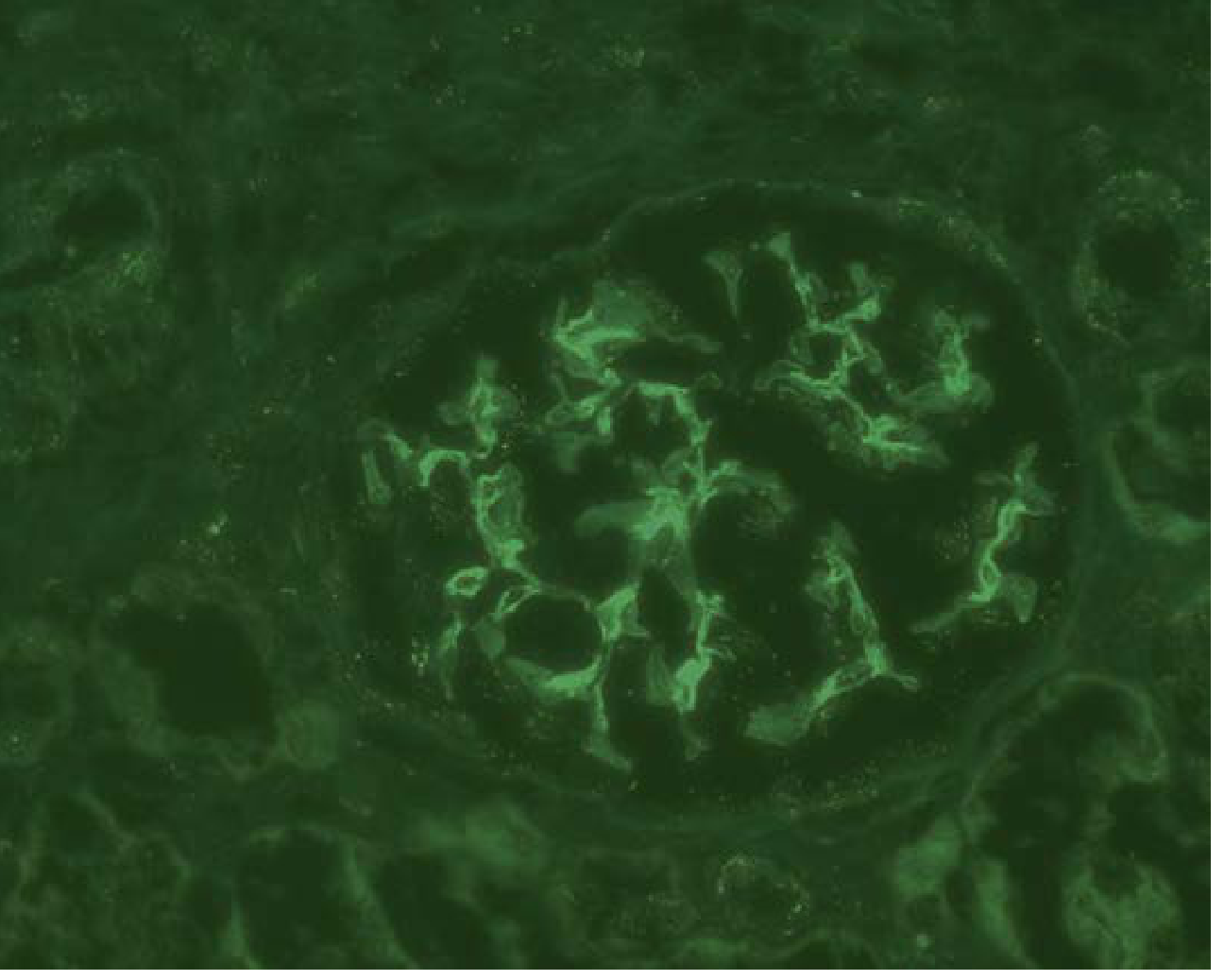
Class IV - Diffuse proliferative LN (H&E showing endocapillary hypercellularity):
Class IV - IgG immunofluorescence (diffuse mesangial + capillary loop staining):
Class V - Glomerulus with membranous thickening (H&E):
Clinical Features
Manifestations vary by histologic class:
- Hematuria (usually microscopic)
- Proteinuria - ranges from subnephrotic to nephrotic-range
- Nephrotic syndrome - especially in class V
- Urinary red cell casts - suggest active proliferative disease
- Renal insufficiency - more common in class III/IV
- Tubular defects
Serological markers of active LN: elevated anti-dsDNA antibodies, low complement (C3, C4).
Diagnosis
ACR criteria (1997) - 4 of 11 required (96% sensitivity and specificity for SLE):
Malar rash, discoid rash, photosensitivity, oral ulcers, non-erosive arthritis, pleuritis/pericarditis, renal disorder, neurologic disorder, hematologic disorder, immunologic disorder (anti-dsDNA, anti-Sm, antiphospholipid antibodies), positive ANA.
SLICC classification (2012) - more sensitive, especially in early SLE:
Requires ≥4 criteria (≥1 clinical + ≥1 immunologic), OR biopsy-proven LN in the presence of ANA or anti-dsDNA (standalone criterion). More sensitive than ACR but slightly less specific.
Kidney biopsy is mandatory for definitive class assignment, which directly guides treatment intensity.
- Henry's Clinical Diagnosis and Management, pp. 1260-1261
Treatment
General Principles
- All patients with LN should receive hydroxychloroquine (reduces flares, improves renal outcomes, safe long-term)
- Treatment is in two phases: induction (rapid disease control) followed by long-term maintenance
- ACE inhibitors or ARBs for proteinuria reduction (hold before conception)
Induction (Classes III/IV, and V with proteinuria):
Two equally acceptable regimens:
- Corticosteroids + IV cyclophosphamide (CYC) - NIH protocol or Euro-Lupus low-dose protocol
- Corticosteroids + mycophenolate mofetil (MMF) - preferred in many centers due to comparable efficacy with better tolerability; treatment evolved from cyclophosphamide-dominant to MMF-dominant regimens since 1980s
Newer agents:
- Belimumab (anti-BLYS) and voclosporin (calcineurin inhibitor) - approved as add-on to MMF + corticosteroids for active LN
- Rituximab (anti-CD20) - evidence suggests it offers no additional benefit over conventional therapy in standard cases; used in refractory disease
Maintenance:
- MMF or azathioprine with low-dose corticosteroids
- Hydroxychloroquine continued long-term
- Duration typically ≥3 years
Therapeutic Plasma Exchange (TPE):
- Large RCT (Lupus Nephritis Collaborative Study Group) showed TPE does not improve clinical outcomes in LN despite more rapid reduction of anti-dsDNA antibodies
- TPE may be considered in select patients with LN + severe thrombotic microangiopathy or refractory antiphospholipid syndrome (American Society for Apheresis: Category III indication)
- Brenner and Rector's The Kidney; Comprehensive Clinical Nephrology 7e
Pregnancy and Lupus Nephritis
- LN flares occur in ~30% of pregnancies and ~15% postpartum
- Prophylactic postpartum corticosteroid dose increase is not indicated
- All pregnant SLE women should continue hydroxychloroquine (reduces flares and fetal growth restriction)
- Safe in pregnancy: corticosteroids, azathioprine, tacrolimus, hydroxychloroquine, low-dose aspirin, LMWH, IVIG, plasma exchange
- Contraindicated in pregnancy: MMF (teratogenic - microtia, cleft palate, auditory canal atresia, micrognathia), cyclophosphamide (teratogenic), rituximab (B-cell depletion in fetus)
- Ideal: disease remission for ≥6 months before conception; MMF switched to azathioprine before conception
- Distinguishing LN flare from preeclampsia is a major challenge (both cause hypertension, proteinuria, AKI, thrombocytopenia); complement levels and anti-dsDNA titers can help
Predictors of poor pregnancy outcome: creatinine >0.9 mg/dL, proteinuria >0.5 g/24h, antiphospholipid syndrome, hypertension, non-White ethnicity, lupus anticoagulant, active disease at conception.
- Comprehensive Clinical Nephrology 7e, p. 645; NKF Primer, p. 495
Prognosis
- High baseline serum creatinine and significant chronic damage on biopsy indicate unfavorable long-term renal prognosis
- Vascular/endothelial abnormalities and crescents on biopsy = aggressive disease
- Anti-Sm antibody (detected in ~49% of biopsy-proven LN in Korea) associated with worse renal prognosis
- ESKD is a major long-term risk, especially in Asian populations
Recent Evidence (PubMed 2024-2026)
A 2024 Brazilian consensus guideline (PMID 38890752) provides updated recommendations on LN diagnosis and treatment, incorporating voclosporin and belimumab as add-on therapies. A 2025 systematic review of LN in Africa (PMID 41057850) highlights late presentation and underdiagnosis in resource-limited settings as major outcome drivers. These are broadly consistent with the textbook framework above.
Sle
Reading File
Finding Sources
Searching PubMed
"systemic lupus erythematosus"[MeSH Terms] AND ("treatment"[MeSH Terms] OR "diagnosis")
Finding Sources
Finding Sources
Searching PubMed
"systemic lupus erythematosus"[MeSH Terms] AND "guideline"
Reading File
Reading File
Reading File
Searching PubMed
"lupus erythematosus, systemic"[MeSH Terms] AND "therapy"[MeSH Subheading]
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Loading Image
Loading Image
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
I now have comprehensive information from Harrison's (2025), Robbins, Fitzpatrick's, Andrews', and Bradley & Daroff's Neurology. I have enough to write a thorough, well-structured answer.
Systemic Lupus Erythematosus (SLE)
SLE is a chronic, multisystem autoimmune disease driven by loss of self-tolerance, leading to autoantibody production against nuclear antigens and widespread immune complex-mediated tissue damage. It predominantly affects young women and follows a relapsing-remitting course.
Epidemiology
- Predominantly affects women of childbearing age (F:M ratio ~9:1)
- Prevalence is highest in Black, Hispanic, and Asian women compared to White women
- Onset typically between ages 15-45; can occur at any age
- Low socioeconomic status, environmental toxins (mercury, pesticides), certain viral infections, and UV light exposure are associated with disease development
Pathogenesis
The central defect is failure to maintain self-tolerance to nuclear antigens, driven by a combination of susceptibility genes, immune dysregulation, and environmental triggers.
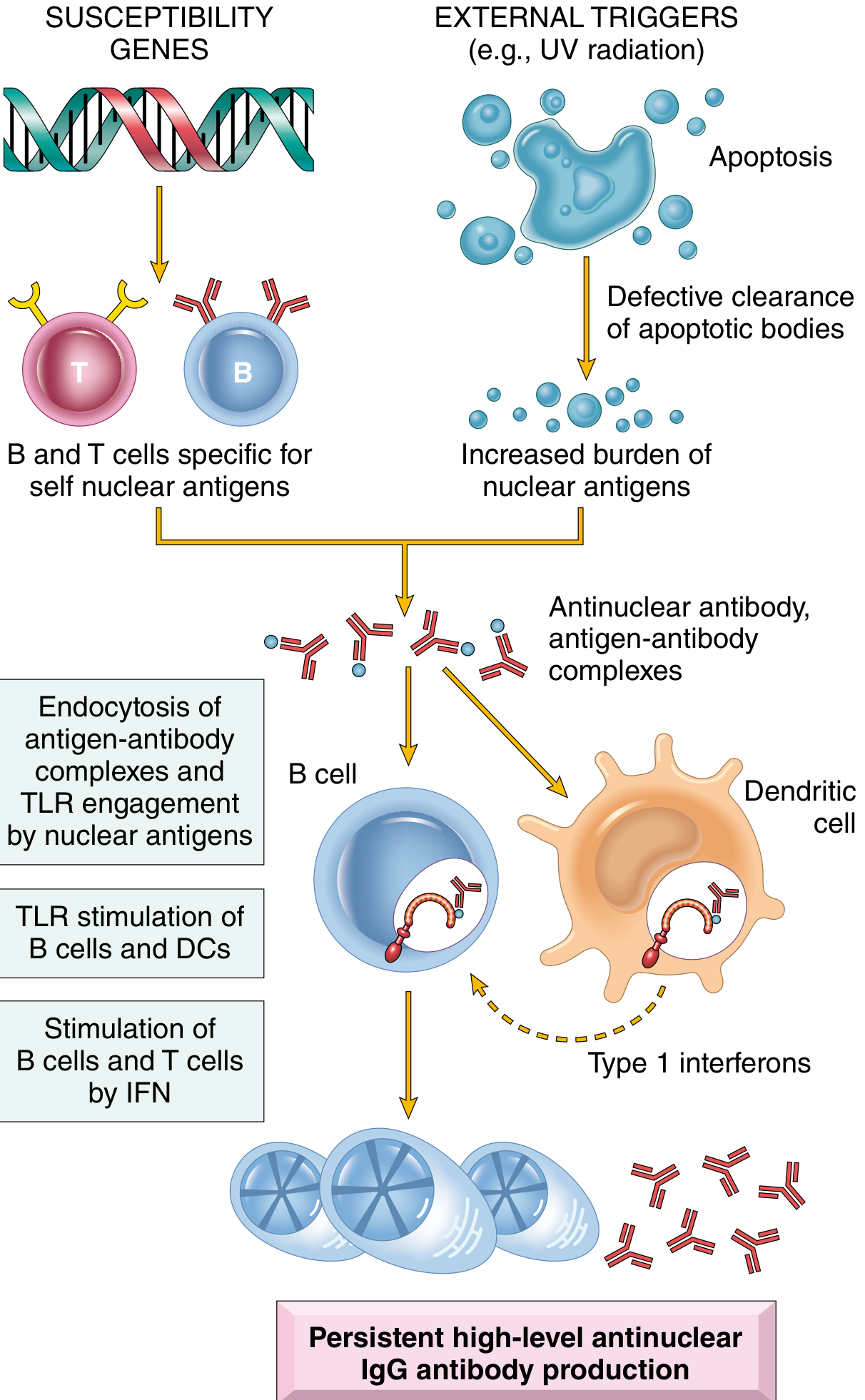
Genetic Factors
- ~25% concordance in monozygotic twins (implying strong environmental contribution too)
- Susceptibility genes impair maintenance of self-tolerance
Environmental Triggers
- UV radiation - alters DNA methylation, generates self-stimulatory nucleic acids, activates keratinocyte immune responses
- EBV infection - induces interferon response, activates SLE susceptibility genes
- Gut microbiome - increased gut permeability promotes translocation of gut microbes and development of lupus-specific autoantibodies
Innate Immunity Defects
- Neutrophils have higher turnover in SLE, delivering a large load of stimulatory nucleic acids
- Defective phagocyte clearance of apoptotic debris and immune complexes
- Toll-like receptors (TLRs) in plasmacytoid dendritic cells are triggered by nucleic acid debris → produce type 1 interferons (IFN-α)
- Type 1 IFN amplifies the autoimmune response by priming neutrophils and sensitizing the adaptive immune system
Adaptive Immunity Defects
- B cells lose tolerance and differentiate (via age-associated B cells, or ABCs) into autoantibody-secreting plasma cells
- T follicular and T peripheral helper cells promote B-cell differentiation into high-affinity autoantibody-secreting plasma cells
- T regulatory cells are defective - fail to maintain tolerance in T and B cells
- Autoantibodies complexed with nucleic acids induce more type 1 IFN in a feed-forward loop
- Harrison's Principles of Internal Medicine 22E (2025), pp. 2871-2872
Autoantibody Profile
| Autoantibody | Sensitivity | Specificity | Clinical Notes |
|---|---|---|---|
| ANA | ~99% | Low | Most sensitive; positive in many other conditions |
| Anti-dsDNA | 70% | High (95%+) | Correlates with disease activity and nephritis; levels rise with flares |
| Anti-Sm | 25-30% | Very high (>99%) | Highly specific for SLE; associated with worse renal prognosis |
| Anti-Ro/SS-A | 30-40% | Moderate | SCLE, neonatal lupus, congenital heart block; also in Sjögren's |
| Anti-La/SS-B | 15-20% | Moderate | Often with anti-Ro |
| Anti-histone | 70% | Low | Strongly positive in drug-induced lupus |
| Antiphospholipid (aPL) | 30-40% | - | Thrombosis, recurrent miscarriage, stroke (antiphospholipid syndrome) |
| Anti-C1q | - | - | Associated with lupus nephritis activity |
- Fitzpatrick's Dermatology; Harrison's; Bradley & Daroff's Neurology
Diagnostic Criteria
ACR Criteria (1997) - 4 of 11 required (96% sensitivity and specificity):
- Malar (butterfly) rash
- Discoid rash
- Photosensitivity
- Oral ulcers
- Non-erosive arthritis
- Serositis (pleuritis or pericarditis)
- Renal disorder (proteinuria >0.5 g/day or casts)
- Neurologic disorder (seizures or psychosis)
- Hematologic disorder (hemolytic anemia, leukopenia, thrombocytopenia)
- Immunologic disorder (anti-dsDNA, anti-Sm, antiphospholipid antibodies)
- Positive ANA
SLICC Criteria (2012) - more sensitive, especially in early SLE:
- Requires ≥4 criteria (≥1 clinical + ≥1 immunologic), OR biopsy-proven lupus nephritis + ANA or anti-dsDNA
- More sensitive than ACR but slightly less specific
- Andrews' Diseases of the Skin; Henry's Clinical Diagnosis
Clinical Manifestations
Systemic / Constitutional
- Fatigue (80-100%), fever (55-85%), weight loss (60%), anorexia
- ~15% have relatively mild disease with fatigue and arthralgia only
Musculoskeletal (80-90%)
- Non-erosive polyarthritis or arthralgia - most common presenting symptom
- Jaccoud's arthropathy (reducible deformities from joint laxity, not erosion)
- Myalgia, myositis
Cutaneous (85%) - 4 of 11 ACR criteria are mucocutaneous
Acute cutaneous LE (ACLE):
- Classic butterfly (malar) rash - erythema on cheeks and bridge of nose, nasolabial fold typically spared (unlike dermatomyositis); resolves without scarring
- Widespread photosensitive erythema
- Interface dermatitis on biopsy
Subacute cutaneous LE (SCLE):
- Annular or psoriasiform plaques, photosensitive; no scarring
- Strongly associated with anti-Ro/SS-A antibodies (70-90%)
Discoid LE (DLE):
- Coin-shaped plaques with dyspigmentation and scarring
- "Carpet-tack" adherent scale in conchal bowl of ear is characteristic
- <5% of DLE patients have the high-titer ANA levels of SLE
Other cutaneous findings:
- Bullous LE (HLA-DR2+; responds dramatically to dapsone; anti-type VII collagen antibodies)
- "Lupus hairs" - short frontal hairs from telogen effluvium and increased fragility
- Diffuse non-scarring alopecia
- Vascular lesions (fingertip/toe edema, nailfold telangiectasia, Raynaud's) in 50%
- Oral/mucosal ulcers (20-30%)
- Livedo reticularis, leg ulcers (suggest antiphospholipid syndrome)
- Lupus panniculitis/profundus - firm nodules with overlying saucer-shaped depressions
- Andrews' Diseases of the Skin; Fitzpatrick's Dermatology; Brenner & Rector
Diagnostic approach flowchart for cutaneous LE:
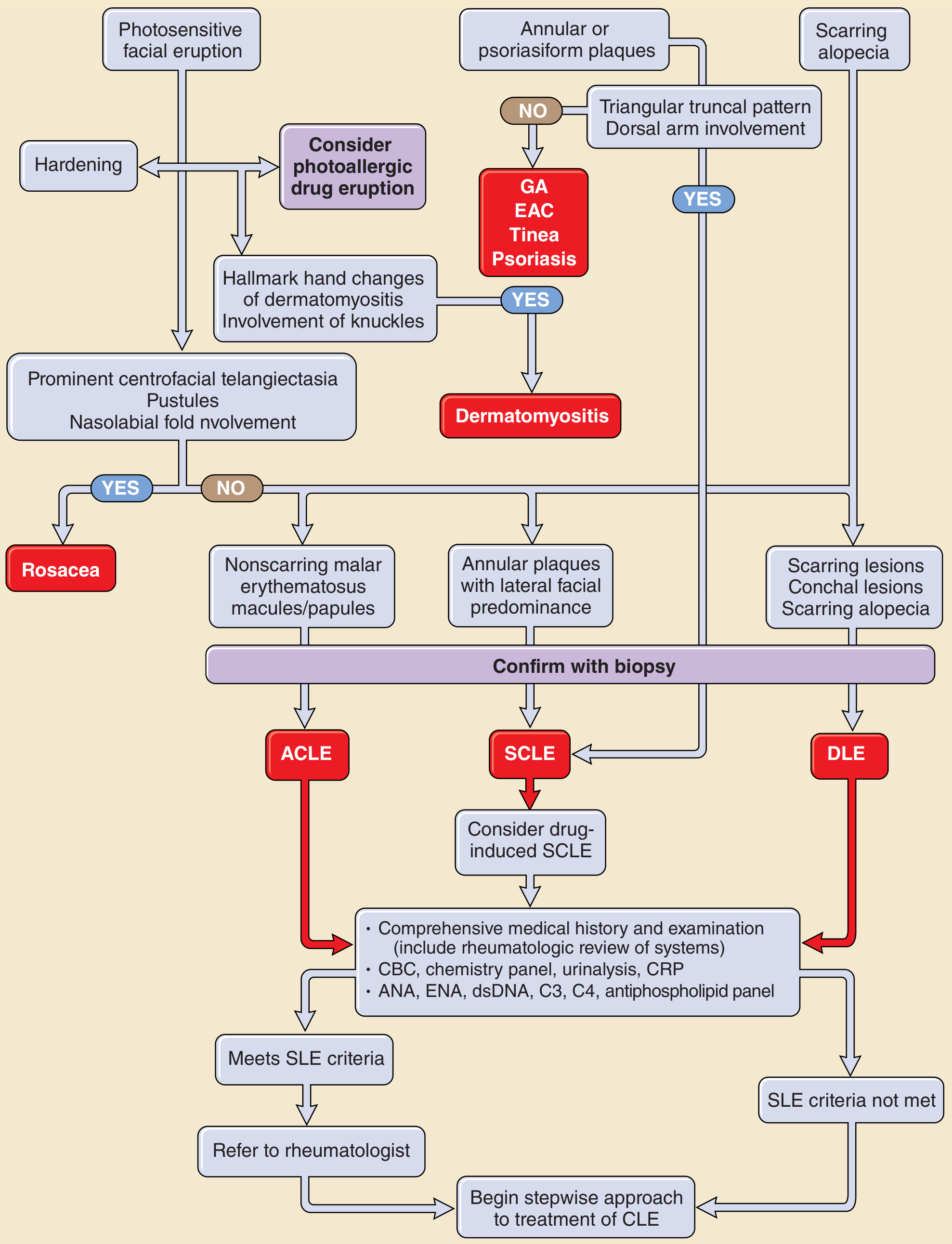
Renal (50-70%)
- Lupus nephritis is the most serious complication (see [prior detailed LN summary])
- Hematuria, proteinuria, casts, renal insufficiency
- Up to 40% have clinically significant renal involvement (glomerulonephritis, tubulointerstitial nephritis)
- ISN/RPS Classification Classes I-VI
Neuropsychiatric SLE (NPSLE) - up to 90% have CNS involvement at some point
- Depression and anxiety each in ~25%
- Cognitive impairment (subclinical) in 11-54%; features include slowed processing, attention deficits, memory impairment
- Seizures - prevalence 3-51%
- Psychosis - 5-8% (but ~30% on prednisone 60-100 mg/day develop psychotic symptoms)
- Cerebrovascular disease - 5-18%; multi-infarct dementia possible
- ANA titer ≥1:40 in up to 99% of SLE patients at some point
- Mechanisms: antineuronal antibodies crossing BBB, antiphospholipid antibodies, inflammatory cytokines
- Bradley and Daroff's Neurology in Clinical Practice
Cardiovascular
- Pericarditis (most common cardiac manifestation) - symptomatic or asymptomatic in up to 50%
- Libman-Sacks endocarditis - nonbacterial verrucous vegetations on either surface of valve leaflets (unlike rheumatic fever which affects lines of closure only); mitral and aortic valves most common
- Accelerated coronary atherosclerosis (especially with long-standing disease and corticosteroid use)
- Myocarditis (less common)
Pulmonary
- Pleuritis/pleural effusions (~50%)
- Lupus pneumonitis, diffuse alveolar hemorrhage (rare but severe)
- Chronic interstitial fibrosis and secondary pulmonary hypertension
Hematologic (100%)
- Anemia (~50%) - normochromic normocytic (chronic disease), autoimmune hemolytic anemia, iron deficiency
- Leukopenia (<4000/μL) in ~50% - usually lymphopenia (<1500/μL) rather than neutropenia
- Thrombocytopenia (<150,000/μL) - usually mild, but can be severe
- Splenomegaly, "onion-skin" penicillary arteries (concentric intimal hyperplasia) in spleen
- Rarely atypical hemolytic-uremic syndrome (schistocytes, low ADAMTS13, elevated LDH)
Gastrointestinal
- Nausea, vomiting, diarrhea during flares
- Mesenteric vasculitis - can cause intestinal perforation, bleeding, ischemia; requires high-dose glucocorticoids
- Elevated liver enzymes (flare, medications, or co-existing autoimmune hepatitis)
- Protein-losing enteropathy
Ocular
- Keratoconjunctivitis sicca (even without secondary Sjögren's)
- Retinal vasculitis (rare; requires aggressive immunosuppression to prevent blindness)
- Episcleritis, scleritis, uveitis, optic neuritis
Antiphospholipid Syndrome (secondary APS)
- Venous and arterial thromboses
- Recurrent spontaneous miscarriages
- Focal cerebral or ocular ischemia
- Associated with IgG/IgM anticardiolipin antibodies and lupus anticoagulant
Morphology (Robbins Pathologic Basis of Disease)
| Organ | Pathological Finding |
|---|---|
| Blood vessels | Acute fibrinoid necrotizing vasculitis → chronic fibrous thickening |
| Kidney | Glomerulonephritis, tubulointerstitial nephritis |
| Skin | Vacuolar degeneration of basal epidermis; perivascular lymphocytic infiltrate; Ig+complement at DEJ on DIF |
| Joints | Non-erosive synovitis |
| CNS | Non-inflammatory vascular occlusion by intimal proliferation |
| Pericardium | Fibrinous pericarditis → fibrous obliteration |
| Heart valves | Libman-Sacks endocarditis - 1-3 mm verrucous deposits on either surface of leaflets |
| Spleen | Onion-skin concentric intimal hyperplasia of penicillary arteries |
| Lungs | Pleuritis, interstitial fibrosis, pulmonary hypertension |
| Bone marrow | LE (hematoxylin) bodies - strongly indicative of SLE |
Treatment
Treatment is stratified by disease severity (SLEDAI score, BILAG classification).
All patients - Background therapy:
- Hydroxychloroquine (HCQ) - backbone of all SLE treatment; reduces flare frequency, improves survival, safe long-term including in pregnancy; max 5 mg/kg/day (retinal toxicity monitoring required)
- Sun protection
- ACE inhibitors/ARBs for renal protection
Mild disease (SLEDAI ≤6, BILAG C):
- NSAIDs (short-term for arthritis/serositis)
- Low-dose corticosteroids
- Antimalarials (HCQ)
- Methotrexate (skin, joints)
Moderate disease (SLEDAI 7-12, ≥2 BILAG B):
- Moderate-dose corticosteroids
- Methotrexate or azathioprine (joints, skin)
- Mycophenolate mofetil (MMF) (serositis, skin, joints)
- Belimumab (anti-BLyS/BAFF) or anifrolumab (anti-IFN-α receptor) as first-line additions in some moderate/severe extrarenal disease
Severe disease (SLEDAI >12, BILAG A, or major organ-threatening):
- IV methylprednisolone pulses then oral prednisone
- IV cyclophosphamide (CYC) - for severe neuropsychiatric SLE, diffuse alveolar hemorrhage, severe vasculitis
- Belimumab and anifrolumab as add-on for severe extrarenal skin/joint disease (NOT recommended for severe neuropsychiatric SLE)
- Rituximab (anti-CD20) - for refractory cytopenias
- IVIG, plasmapheresis - for severe NPSLE, TTP-like disease
- Anticoagulation (VKA) for APS-related thrombosis
Biological agents (summary):
| Drug | Target | Indication |
|---|---|---|
| Belimumab | BLyS/BAFF | Extrarenal SLE, LN (add-on) |
| Anifrolumab | IFN-α receptor | Extrarenal SLE (skin, joints); NOT for severe NPSLE |
| Rituximab | CD20 | Refractory disease, severe cytopenias |
| Voclosporin | Calcineurin | Lupus nephritis (add-on to MMF) |
| Telitacicept | BLyS + APRIL | Phase 3 RCT positive (2025 NEJM) |
- Harrison's Principles of Internal Medicine 22E (2025), p. 2877; Braunwald's Heart Disease
Recent Evidence (PubMed 2025-2026)
- 2025 EULAR Guidelines (PMID 41107121) - updated recommendations for SLE with kidney involvement, endorsing belimumab and voclosporin as add-on therapies for LN
- Telitacicept Phase 3 RCT (PMID 41092329, NEJM Oct 2025) - telitacicept (dual BLyS + APRIL inhibitor) showed significant benefit in SLE; a new emerging biologic option
- Cutaneous LE review (PMID 41204012, Nat Rev Rheumatol Dec 2025) - updated pathogenesis-to-targeted-therapy review
Prognosis & Monitoring
- Goals: low disease activity or remission on ongoing therapy
- Majority of patients have active disease despite therapy or frequent flares
- Major causes of morbidity: renal failure (LN), cardiovascular disease (accelerated atherosclerosis), infections (from immunosuppression)
- Monitoring: Anti-dsDNA titers, complement (C3, C4), CBC, urinalysis with microscopy, creatinine, BP
- HCQ retinal toxicity screen annually after 5 years of use
- Anti-Ro/SS-A screening in all pregnant women (neonatal heart block risk)
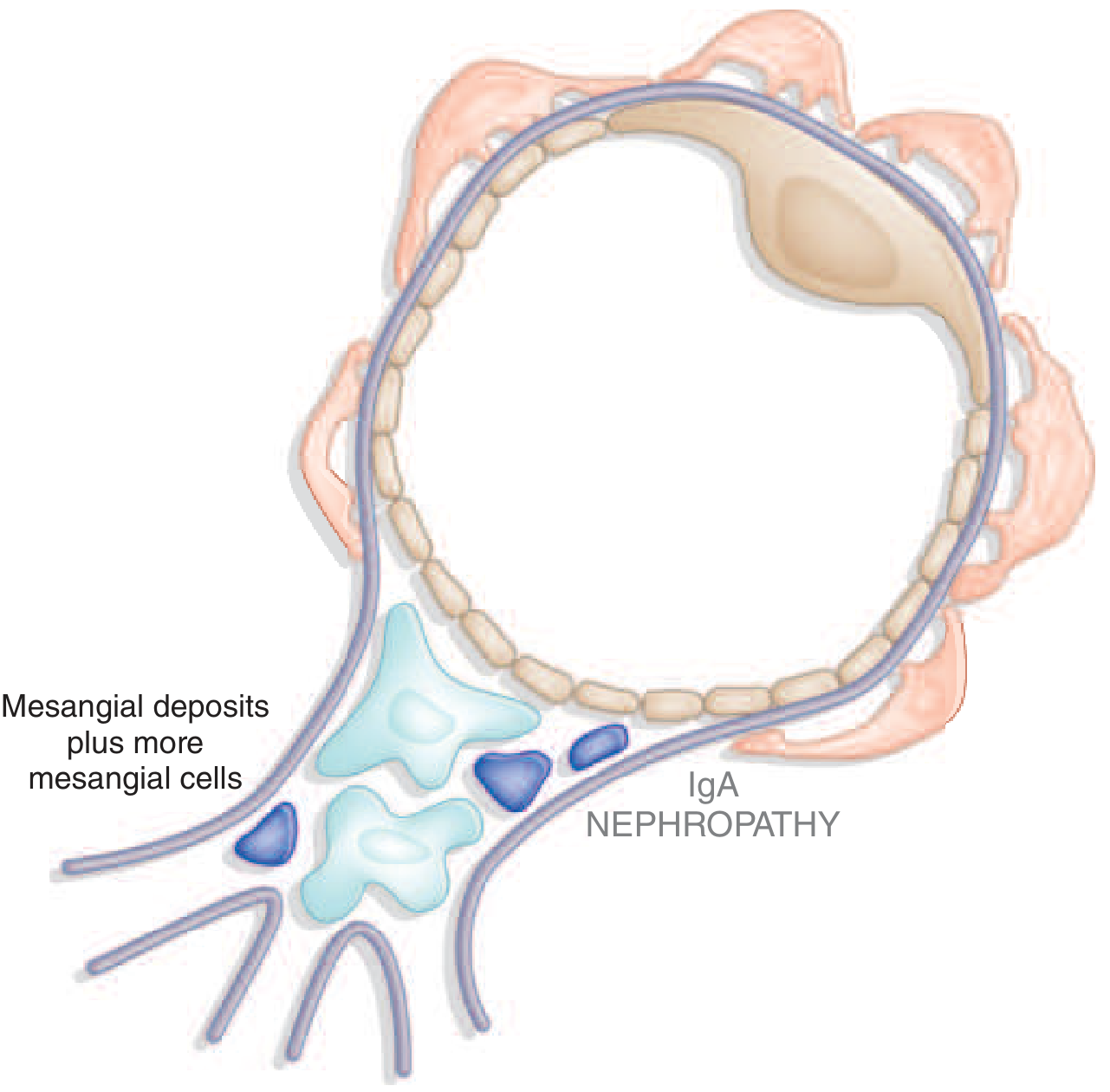
IgA nephropathy
Reading File
Finding Sources
Searching PubMed
"IgA nephropathy"[MeSH Terms] AND "therapy"[MeSH Subheading]
Reading File
Reading File
Reading File
Searching PubMed
"glomerulonephritis, IGA"[MeSH Terms] AND treatment
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
I now have comprehensive material covering pathogenesis (four-hit model), MEST-C scoring, clinical features, treatment, and prognosis. I have two landmark 2025 NEJM trials. Let me compose the full answer.
IgA Nephropathy (Berger Disease)
IgA nephropathy (IgAN) is the most common primary glomerulonephritis worldwide. It is defined by dominant or co-dominant mesangial IgA1 deposits on kidney biopsy and carries a real risk of progression to ESKD in a substantial proportion of patients.
Epidemiology
- Most common primary glomerulonephritis globally - accounts for 40-50% of primary GN in Asia (>45% of primary GN in China) and 10-20% in the USA
- Peak incidence: 2nd and 3rd decades of life
- Male predominance in White populations (M:F ratio 2:1 to 3:1); approaches 1:1 in Asian populations
- Very uncommon in Black populations
- Rare familial clustering; predominantly sporadic
- Geographic variation in prevalence likely reflects both true differences and differences in detection (biopsy thresholds)
- Henry's Clinical Diagnosis and Management; Comprehensive Clinical Nephrology 7e; Harrison's 22E
Pathogenesis - The Four-Hit Model
IgAN is an autoimmune disease driven by aberrant IgA1 glycosylation. The currently accepted model proposes four sequential "hits":
| Hit | Event |
|---|---|
| 1 | Aberrant O-glycosylation of IgA1 - galactose-deficient IgA1 (Gd-IgA1) is produced in excess, especially by B cells responding to mucosal infections (tonsillitis, URI, GI infections). Instead of galactose, the hinge region O-linked glycans contain N-acetylgalactosamine |
| 2 | Autoantibody recognition - IgG (and sometimes IgA) autoantibodies recognize the exposed N-acetylgalactosamine on Gd-IgA1 as foreign and bind to it |
| 3 | Immune complex formation - Gd-IgA1-containing immune complexes form in the circulation; anti-IgA1 autoantibodies have somatic mutations suggesting antigen-driven affinity maturation |
| 4 | Mesangial deposition and injury - Immune complexes deposit in the glomerular mesangium, activate mesangial cells → proliferation, increased extracellular matrix, cytokine/chemokine production → parenchymal injury and fibrosis |
Complement activation occurs via the lectin and alternative pathways (NOT the classical pathway - C1q is characteristically absent). Properdin co-deposits in 75-100% of cases, and C5b-9 (terminal complement complex) deposits are common. This is a key diagnostic distinction from other immune complex GNs.
Mucosal infections (especially upper respiratory and GI) trigger increased mucosal IgA production → more Gd-IgA1 → nephritogenic immune complex formation → disease flares.
Genetic susceptibility loci include genes involved in antigen processing, MHC, the DEFA gene cluster (mucosal defense), and alternative complement pathway components.
- Robbins & Kumar Basic Pathology; Henry's; Brenner & Rector; Comprehensive Clinical Nephrology 7e
Diseases Associated with Secondary IgAN
Mesangial IgA deposits can occur in systemic diseases but are not usually associated with clinically significant kidney disease in those settings:
| Disease Group | Associations |
|---|---|
| Rheumatic/autoimmune | Ankylosing spondylitis, rheumatoid arthritis, Reiter syndrome, uveitis |
| GI disease | Celiac disease, Crohn disease, ulcerative colitis |
| Hepatic disease | Alcoholic liver disease, nonalcoholic cirrhosis (impaired hepatobiliary clearance of IgA complexes) |
| Skin | Dermatitis herpetiformis |
| Pulmonary | Sarcoidosis |
| Infections | HIV, chronic bronchiectasis; IgA-dominant Staphylococcus-associated GN - associated with clinically significant kidney disease |
| Immunologic | IgA monoclonal gammopathy, Sjögren syndrome |
| IgA vasculitis (IgAV) | Formerly Henoch-Schönlein purpura - same glomerular lesion but with systemic vasculitis, palpable purpura, arthritis, abdominal pain; younger age (<20 years); more capillary loop IgA deposits |
- Comprehensive Clinical Nephrology 7e, p. 333; Harrison's 22E
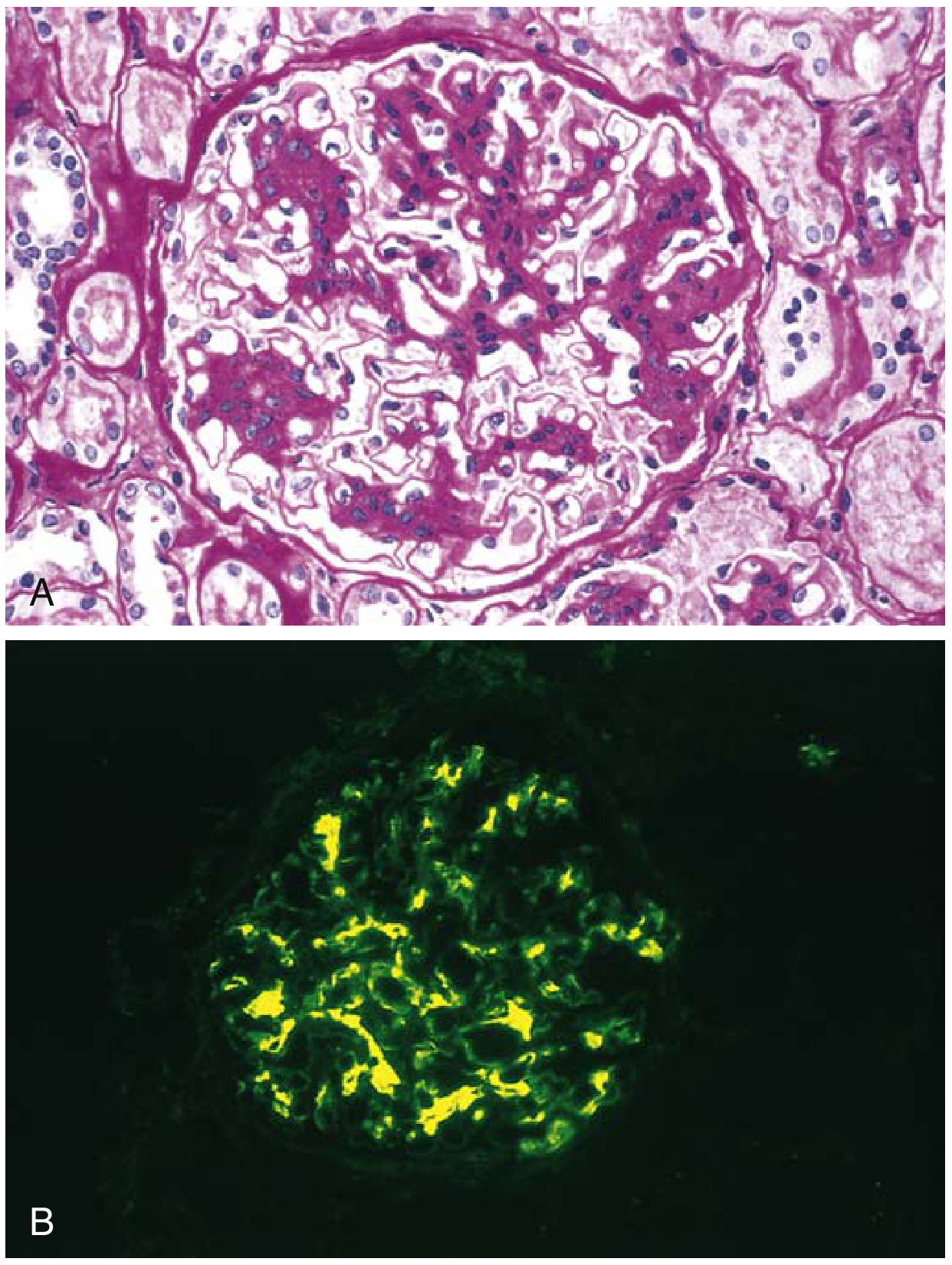
Biopsy - Histological Findings
Immunofluorescence (diagnostic hallmark)
- Dominant or codominant mesangial IgA deposition (must exceed any other immunoglobulin)
- Typically polymeric IgA1 with J-chain
- C3 co-deposits in up to 90%
- IgG in ~40%, IgM in ~40%
- C1q absent (distinguishes from lupus nephritis which shows "full house")
- Properdin (alternative pathway) + factor H commonly co-deposit
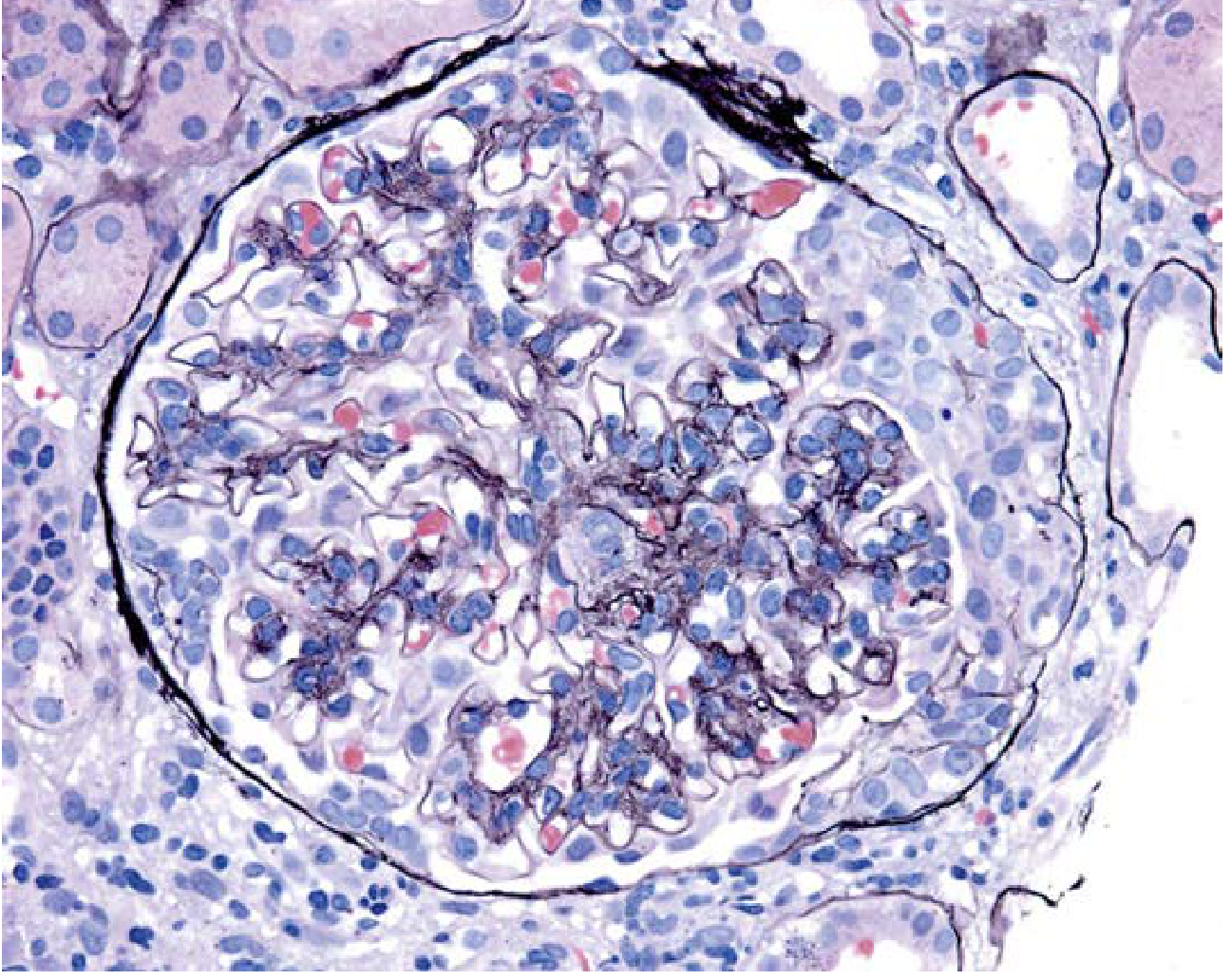
Light Microscopy
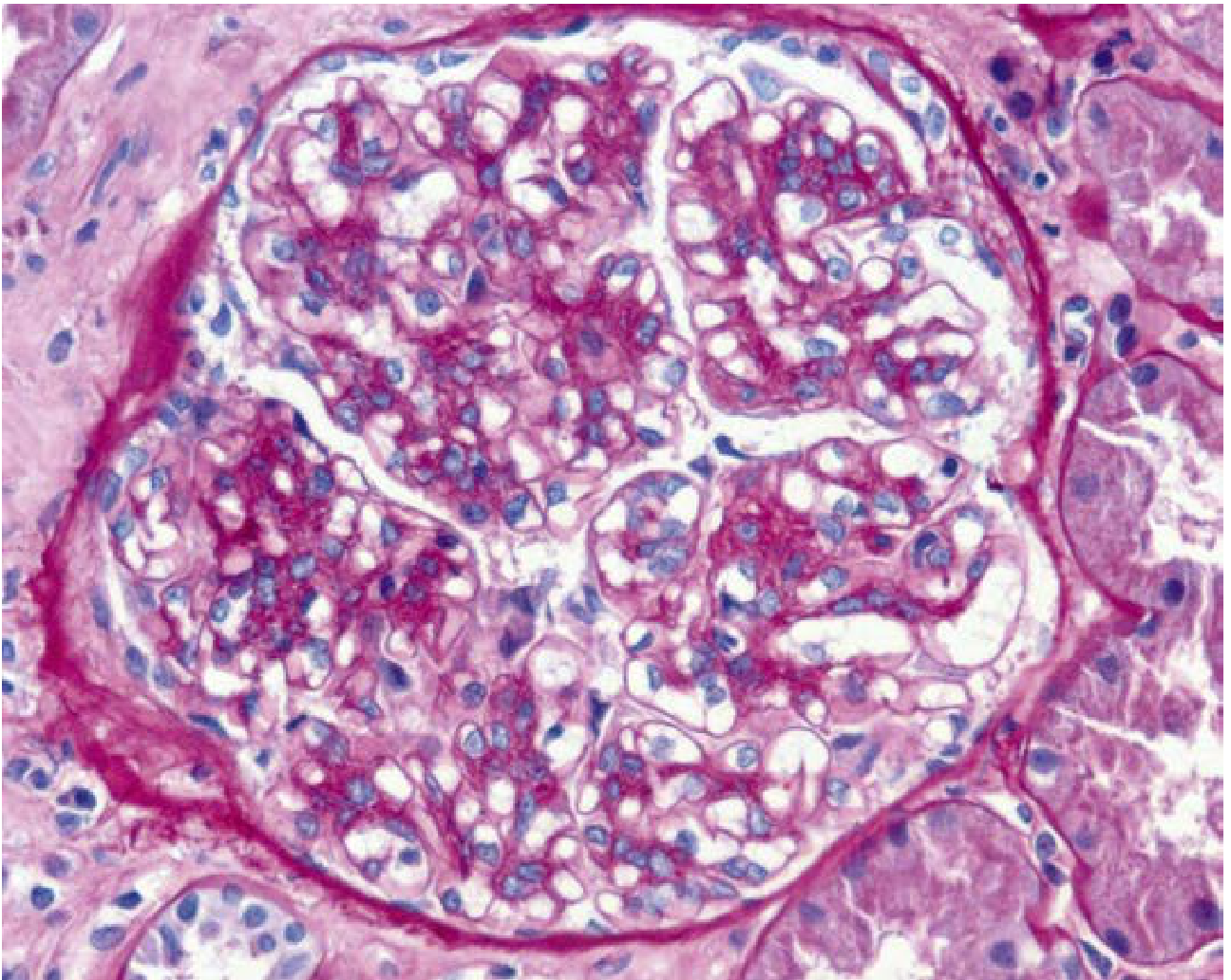
Variable appearance - the hallmark is mesangial hypercellularity and matrix expansion:
- Mesangial widening and hypercellularity (most common)
- Endocapillary hypercellularity
- Focal segmental sclerosis (indicates chronicity)
- Crescents (aggressive disease; RPGN presentation)
- Rarely, nearly normal glomeruli (mild disease)
Electron Microscopy
- Mesangial and paramesangial electron-dense deposits (confirming the IF pattern)
- Subepithelial/subendothelial deposits also possible
- Capillary loop deposits indicate worse prognosis
Key Histological Images
IgA nephropathy - LM (A) and IgA immunofluorescence (B):
IgA nephropathy with crescent formation (aggressive disease):
IgA nephropathy diagram - mesangial deposits:
Mesangial hypercellularity with matrix increase (PAS, x400):
Oxford (MEST-C) Classification
The Oxford Classification identifies five histologic features with independent prognostic value, scorable reproducibly on LM:
| Feature | Score | Definition |
|---|---|---|
| M - Mesangial hypercellularity | M0 / M1 | >3 mesangial cells per mesangial area in >50% of glomeruli = M1 |
| E - Endocapillary hypercellularity | E0 / E1 | Any glomerulus with endocapillary hypercellularity |
| S - Segmental sclerosis | S0 / S1 | Any glomerulus with segmental sclerosis or adhesion; S1 can be refined for tip lesions (associated with proteinuria) |
| T - Tubular atrophy/Interstitial fibrosis | T0 / T1 / T2 | 0-25% (T0), 26-50% (T1), >50% (T2) of cortical area |
| C - Crescents | C0 / C1 / C2 | No crescents (C0), crescents in <25% of glomeruli (C1), ≥25% (C2) |
Higher scores (especially T and C scores) predict worse renal outcomes. The MEST-C score maintains predictive value even at long-term follow-up.
- Comprehensive Clinical Nephrology 7e; Henry's Clinical Diagnosis
Clinical Presentations
| Presentation | Frequency | Notes |
|---|---|---|
| Episodic macroscopic hematuria | 40-50% | Most common under age 40; brown urine, not red; clots unusual; occurs within 24 hours of mucosal infection (vs. 2-3 week delay in post-streptococcal GN); termed "synpharyngitic hematuria" |
| Asymptomatic microscopic hematuria ± proteinuria | 30-40% | Usually proteinuria <2 g/24h; found on screening |
| Persistent proteinuria with hematuria | 15-20% | May have nephrotic-range proteinuria |
| Nephrotic syndrome | <10% | Less common; consider concurrent minimal change disease |
| Rapidly progressive GN (RPGN) | Rare | Crescentic IgAN; aggressive disease |
| Chronic renal failure | - | Late presentation; older age; worse prognosis |
Episodes of macroscopic hematuria may be associated with acute kidney injury from tubular occlusion by RBCs - usually reversible. Macroscopic hematuria itself does not worsen long-term prognosis.
Diagnosis
- Kidney biopsy is required - no blood or urine biomarker is diagnostic
- Serum IgA levels are elevated in 20-50% of patients, but are not diagnostic
- Serum IgA deposits found in skin biopsies in 15-55% of patients
- Biopsy confirms dominant mesangial IgA1 on IF
Prognosis
- 20-40% of patients reach ESKD over 20-30 years (varies considerably by population and risk factors)
- Median life expectancy reduced by ~10 years in some populations (USA southeastern cohort)
- Episodes of macroscopic hematuria alone do NOT confer worse prognosis
- Older age at diagnosis is associated with worse prognosis (likely reflects later stage disease)
Poor prognostic factors (Box 24.2 - IgAN Risk Prediction):
- Persistent proteinuria (time-averaged proteinuria is the strongest modifiable predictor; risk is low when <0.2 g/24h)
- Hypertension at presentation
- Reduced eGFR at biopsy
- Higher MEST-C scores (especially T1/T2 and C2)
- Capillary loop IgA deposits on IF
- Hyperuricemia, smoking, high BMI
- Persistent hematuria (hematuria remission associated with better outcomes)
- Comprehensive Clinical Nephrology 7e, p. 336
Treatment
Supportive / Baseline Therapy (all patients):
- ACE inhibitors or ARBs - cornerstone for all patients with proteinuria >0.5-1 g/day; reduce proteinuria and slow progression; target BP <130/80 mmHg
- SGLT2 inhibitors (e.g., dapagliflozin) - now shown to reduce adverse kidney outcomes in IgAN; recommended as add-on in patients at risk of progression
- Lifestyle: BP control, smoking cessation, weight management, treatment of hyperuricemia
- Management of underlying conditions (e.g., celiac disease, tonsillitis)
- Tonsillectomy - suggested benefit in select Asian patients with recurrent tonsillitis-triggered hematuria; evidence limited
Immunosuppression (selected patients at high risk of progression):
- Targeted-release budesonide (Nefecon) - oral formulation delivering budesonide to the ileum/Peyer's patches; shown to reduce proteinuria and preserve eGFR in patients at risk of rapid progression (Phase 3 NefIgArd trial); approved by FDA 2023 for IgAN with urine protein-creatinine ratio ≥1.5 g/g
- Systemic corticosteroids - conflicting evidence; the STOP-IgAN and TESTING trials showed benefit in reducing proteinuria but increased serious adverse events; use with caution; some guidelines recommend a 6-month course in high-risk patients
- Fish oil (omega-3 fatty acids) - modest benefit in some small studies; generally safe
- Cytotoxic agents (cyclophosphamide, azathioprine) - reserved for crescentic/RPGN presentation
- Plasmapheresis - considered for RPGN presentation with crescents
RPGN presentation:
- Systemic corticosteroids + cyclophosphamide or rituximab
- Plasmapheresis considered
- Harrison's 22E; Comprehensive Clinical Nephrology 7e
Recent Evidence - Landmark 2025 NEJM Trials
Two Phase 3 RCTs published in NEJM Feb 2025 represent a paradigm shift in IgAN treatment:
1. Iptacopan (Alternative Complement Pathway Inhibitor)
- PMID 39453772 - APPLAUSE-IgAN trial
- Iptacopan is an oral factor B inhibitor (blocks the alternative complement pathway amplification loop)
- Significantly reduced proteinuria vs. placebo in patients with IgAN
- First complement inhibitor approved/in late-stage development specifically targeting the alternative pathway in IgAN
2. Atrasentan (Endothelin-1 Receptor Antagonist)
- PMID 39460694 - ALIGN trial
- Atrasentan is a selective endothelin-A receptor antagonist
- Phase 3 trial showed significant reduction in proteinuria and kidney function preservation in IgAN patients
- Builds on its earlier use in diabetic nephropathy
Additional Recent Evidence:
- Nat Rev Nephrol 2025 (PMID 39232245) - comprehensive pathogenesis-to-treatment review highlighting the multi-hit model and new therapeutic targets
- Kidney Int 2025 (PMID 39894081) - "call for a new paradigm" review supporting combination therapy targeting multiple pathways simultaneously
- IPNA Guidelines 2025 (PMID 39331079) - pediatric IgAN and IgA vasculitis nephritis recommendations
Emerging Therapeutic Targets
The multi-hit pathogenesis model offers several therapeutic windows now being actively studied:
| Target | Drug Example | Mechanism |
|---|---|---|
| Gd-IgA1 production | Targeted-release budesonide (Nefecon) | Reduces Peyer's patch IgA1 production |
| Alternative complement | Iptacopan (factor B inhibitor) | Blocks alternative pathway amplification |
| Lectin complement pathway | Sibeprenlimab (anti-MASP-2) | Blocks lectin pathway |
| Endothelin receptor | Atrasentan | ET-A antagonist, anti-proteinuric |
| B-cell activation | Sparsentan (dual AT1R + endothelin blocker) | Also anti-proteinuric via dual mechanism |
| SGLT2 | Dapagliflozin, empagliflozin | Renoprotective, anti-inflammatory |
This is a shared conversation. Sign in to Orris to start your own chat.