Thalesemia in pediatrics refer op ghai 10th edition
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Searching Images
thalassemia major facies skull x-ray peripheral blood smear
Finding Sources
Reading File
Reading File
Reading File
I now have comprehensive material from multiple authoritative sources. Let me compile a complete pediatric thalassemia review in OP Ghai style.
Note: OP Ghai's Essential Pediatrics (10th edition) is not in the textbook library. The following is a comprehensive, pediatrics-oriented review compiled from Robbins & Cotran Pathologic Basis of Disease, Goldman-Cecil Medicine, Harriet Lane Handbook (23rd ed.), and Emery's Elements of Medical Genetics, covering all topics as presented in Ghai's standard structure.
Thalassemia in Pediatrics
Definition
Thalassemias are a group of inherited disorders caused by mutations that decrease the synthesis of α- or β-globin chains, leading to:
- Hemoglobin deficiency (hypochromic, microcytic anemia)
- Relative excess of the other (normal) globin chain → intracellular precipitation → hemolysis and ineffective erythropoiesis
The name derives from thalassa (Greek for "sea"), reflecting its prevalence in Mediterranean populations.
Epidemiology & Genetics
- Endemic regions: Mediterranean basin, Middle East, tropical Africa, Indian subcontinent, Southeast Asia
- Autosomal codominant inheritance
- Selective advantage: heterozygotes are protected against P. falciparum malaria
- α-globin genes: 2 genes in tandem on chromosome 16 (total 4 alleles)
- β-globin gene: Single gene on chromosome 11
Classification
β-Thalassemia
| Clinical Syndrome | Genotype | Clinical Features |
|---|---|---|
| β-thalassemia major (Cooley's anemia) | β⁰/β⁰, β⁺/β⁰, β⁺/β⁺ | Severe; regular transfusions required |
| β-thalassemia intermedia | Variable | Moderate; transfusions not always required |
| β-thalassemia minor (trait) | β⁺/β, β⁰/β | Asymptomatic; mild or absent anemia |
α-Thalassemia
| Clinical Syndrome | Genes Deleted | Clinical Features |
|---|---|---|
| Silent carrier | 1 gene (-α/αα) | Asymptomatic; no red cell abnormality |
| α-Thalassemia trait | 2 genes (--/αα or -α/-α) | Mild microcytic anemia; like β-minor |
| HbH disease | 3 genes (--/-α) | Moderate-severe hemolytic anemia |
| Hydrops fetalis | 4 genes (--/--) | Lethal in utero (HbBart = γ₄) |
Molecular Pathogenesis of β-Thalassemia
Three major classes of β-globin mutations (>400 known):
- Splicing mutations — most common cause of β⁺; ectopic splice site allows some normal mRNA
- Promoter mutations — reduce transcription ~75–80%; cause β⁺⁺
- Chain terminator mutations — most common cause of β⁰; nonsense or frameshift → no functional β-globin
Consequence of excess free α-chains:
- Precipitate within erythroid precursors → inclusion bodies
- Membrane damage → ineffective erythropoiesis in the marrow
- Surviving red cells: abnormal, short-lived → hemolysis
- Massive erythroid hyperplasia → bone marrow expansion → skeletal deformities
- Erythroferrone ↑ → hepcidin ↓ → ↑ intestinal iron absorption → secondary hemochromatosis
β-Thalassemia Major — Clinical Features in Children
Onset: 6–9 months of age (as HbF→HbA switch occurs)
Symptoms:
- Severe progressive anemia (Hb 3–6 g/dL in untransfused)
- Failure to thrive, growth retardation
- Pallor, jaundice, hepatosplenomegaly (extramedullary hematopoiesis)
Skeletal changes (from marrow hyperplasia):
- "Chipmunk facies" — frontal bossing, maxillary hyperplasia, prominent malar eminences
- "Hair-on-end" (crew-cut) appearance on skull X-ray — perpendicular striations on outer table
- Thinning of cortex, pathological fractures
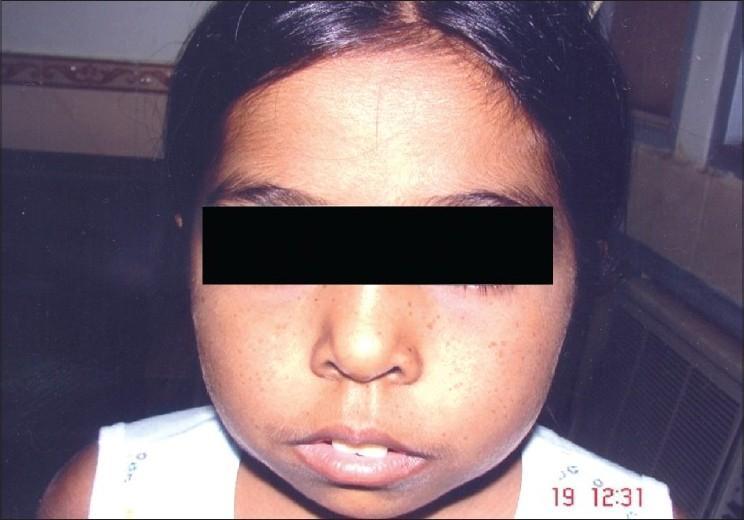
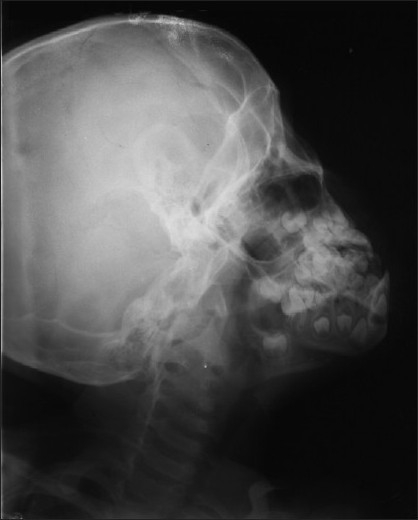
Complications of iron overload (from transfusions + increased gut absorption):
- Cardiac: dilated cardiomyopathy, arrhythmias — leading cause of death
- Hepatic: cirrhosis
- Endocrine: delayed puberty, hypogonadism, diabetes mellitus, hypothyroidism
- Skin: bronze pigmentation (hemosiderosis)
Blood film: Marked anisocytosis, poikilocytosis, microcytosis, hypochromia, target cells, nucleated RBCs, basophilic stippling
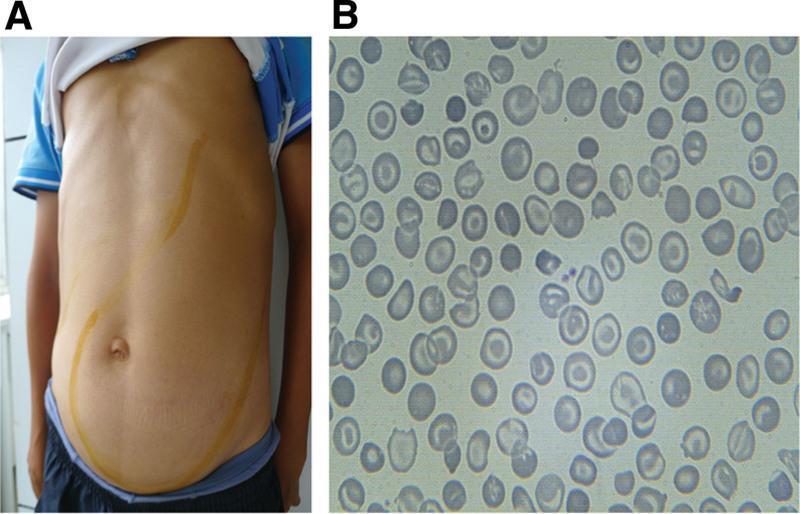
β-Thalassemia Minor (Trait)
- Usually asymptomatic
- Mild microcytic hypochromic anemia
- MCV <80 fL, MCH <27 pg, HbA₂ >3.5% (diagnostic)
- High RBC count (pseudopolycythemia)
- No specific treatment; genetic counseling mandatory
- Folic acid supplementation in pregnancy
α-Thalassemia in Pediatrics
HbH disease (3 genes deleted):
- Hb 8–9 g/dL; does not require regular transfusion
- Reticulocytosis, target cells, basophilic stippling
- Brilliant cresyl blue stain → multiple inclusion bodies (HbH precipitates)
- Folic acid 2–5 mg/day recommended
- Risk: hemolytic crises during infections
Hydrops fetalis (4 genes deleted):
- HbBart (γ₄) has very high O₂ affinity → cannot deliver O₂ → severe fetal hypoxia
- Features: massive organomegaly, heart failure, severe anemia (Hb 3–20 g/dL), generalized edema
- Fatal without intrauterine transfusion
- Maternal risks: retained placenta, eclampsia, sepsis
Diagnosis
CBC & Indices
- ↓ MCV (<80 fL), ↓ MCH (<27 pg), ↑ RBC count
- Normal serum iron/ferritin (to exclude IDA)
Hemoglobin Electrophoresis / HPLC
- β-thalassemia minor: HbA₂ >3.5% (diagnostic); HbF mildly elevated
- β-thalassemia major: HbA markedly ↓ or absent; HbF markedly ↑; HbA₂ variable
- HbH disease: HbA₂ ↓; HbH (β₄) detected
Peripheral Blood Smear
- Microcytosis, hypochromia, anisocytosis, poikilocytosis, target cells, nucleated RBCs, basophilic stippling, tear-drop cells
Bone Marrow (if done)
- Marked erythroid hyperplasia
Molecular Diagnosis
- PCR-based methods (ARMS-PCR, gap-PCR, direct sequencing) — identifies specific mutations
- Multiplex ligation-dependent probe amplification (MLPA) — for α-gene deletions
Management
Blood Transfusion (Thalassemia Major)
- Hypertransfusion regimen: Maintain pre-transfusion Hb >9–10.5 g/dL
- Transfuse every 2–5 weeks with leukoreduced packed red cells
- Initiate when Hb consistently <7 g/dL with failure to thrive or bone changes
- Suppresses ineffective erythropoiesis → prevents skeletal deformity and organomegaly
Iron Chelation Therapy
Initiate after 10–20 transfusions or serum ferritin >1000 μg/L
| Drug | Route | Notes |
|---|---|---|
| Deferoxamine (DFO) | SC/IV infusion (8–12 hrs/day, 5–7 days/week) | Gold standard; risk: local reactions, audiological/visual toxicity |
| Deferiprone | Oral (3× daily) | Safe in young children; risk: agranulocytosis, arthropathy |
| Deferasirox | Oral (once daily) | Preferred oral agent; monitor renal function and LFTs |
Splenectomy
- Indicated if transfusion requirement increases markedly (>1.5× baseline)
- Increases risk of overwhelming sepsis (especially S. pneumoniae, H. influenzae, Neisseria)
- Vaccinate against pneumococcus, meningococcus, Hib 4–6 weeks before splenectomy
- Lifelong penicillin prophylaxis post-splenectomy
Folic Acid
- Supplementation due to chronic hemolysis and ↑ demand
- 5 mg/day recommended
Curative Therapy
- Allogeneic hematopoietic stem cell transplantation (HSCT): Best results in pediatric patients with HLA-identical sibling donors; ideally before significant organ damage
- Gene therapy: Corrective gene therapy with encouraging results; approved in some settings for β-thalassemia
Endocrine & Supportive Care
- Monitor for growth retardation, delayed puberty, hypothyroidism, diabetes
- Hormone replacement as needed; calcium + Vitamin D for bone health
- Hepatitis B vaccination; screen for transfusion-transmitted infections (HBV, HCV, HIV)
Prognosis
- Thalassemia minor: Normal life expectancy
- Thalassemia major (well-managed): Survival now into 3rd–4th decade with regular transfusion + chelation; cardiac failure remains the leading cause of death in poorly chelated patients
- Hydrops fetalis: Fatal without intrauterine transfusion
Prevention & Screening
- Carrier detection: MCV, MCH, HbA₂ in high-risk populations
- Prenatal diagnosis: Chorionic villus sampling (10–12 weeks) or amniocentesis (16 weeks) + PCR-based mutation analysis
- Genetic counseling: Both parents with thalassemia trait → 25% chance of affected child; counseling mandatory before pregnancy
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease (9780443264528) — Goldman-Cecil Medicine, 26e (9780323930345) — Harriet Lane Handbook, 23rd ed. (9780323876988) — Emery's Elements of Medical Genetics (9780702079665)
This is a shared conversation. Sign in to Orris to start your own chat.