Prepare a long essay for pcr
Asking for Preferences
Post graduate Pathology exam
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
Here is a comprehensive, postgraduate-level essay on PCR for your pathology exam:
POLYMERASE CHAIN REACTION (PCR)
Introduction
The Polymerase Chain Reaction (PCR) is a simple but extraordinarily powerful in vitro technique that permits the enzymatic amplification of a specific nucleic acid sequence from a complex mixture of DNA or RNA. Conceived by Kary Mullis in 1983 and first described by Saiki and colleagues in 1985 and 1988, PCR has been described as a milestone in biotechnology that heralded the beginning of the era of molecular diagnostics. It was recognized with the Nobel Prize in Chemistry in 1993. Within a matter of hours, a single copy of a target DNA sequence can be amplified to produce billions of identical copies, enabling detection, analysis, and quantification that were previously impossible. In pathology, PCR has become an indispensable tool, finding application in the diagnosis of infectious diseases, genetic disorders, neoplasms, forensic identification, and the monitoring of minimal residual disease.
Basic Principle and Components
At its core, PCR is a process of repeated enzymatic copying of a defined DNA segment. It exploits the ability of DNA polymerase to synthesize a new strand of DNA complementary to a template strand, provided that a short oligonucleotide primer is already annealed to the template to provide a free 3'-OH group for extension.
Essential components of a PCR reaction include:
- Template DNA - The nucleic acid containing the target sequence to be amplified.
- Two oligonucleotide primers - Typically 18 to 30 nucleotides long. They flank the target sequence and are complementary to opposite strands of the double-stranded DNA template. One primer (forward) is complementary to one strand; the other (reverse) is complementary to the opposite strand.
- Thermostable DNA polymerase - Most commonly Taq polymerase, derived from Thermus aquaticus, a thermophilic bacterium. Its heat stability allows it to survive the high-temperature denaturation steps without losing activity.
- Deoxyribonucleotide triphosphates (dNTPs) - An equimolar mixture of dATP, dCTP, dGTP, and dTTP, the building blocks for new DNA strands.
- Magnesium chloride (MgCl₂) - A divalent cation cofactor essential for Taq polymerase activity; its concentration critically affects specificity and efficiency.
- Buffer - Typically Tris-HCl with KCl, to maintain optimal pH and ionic strength.
The Three Steps of a PCR Cycle
Each cycle of PCR consists of three temperature-dependent steps, carried out in a programmable thermal cycler:
1. Denaturation (94-96°C)
The reaction mixture is heated to approximately 94-96°C. This high temperature disrupts the hydrogen bonds holding the two complementary strands of the double-stranded DNA together, separating them into two single-stranded templates. Sufficient denaturation is critical; incomplete denaturation leads to poor amplification.
2. Annealing (50-65°C)
The temperature is lowered to allow the primers to anneal to their complementary sequences on the single-stranded template DNA. The annealing temperature is typically calculated as approximately 5°C below the melting temperature (Tm) of the primers. Too high a temperature leads to inefficient primer binding (false negatives); too low a temperature allows non-specific primer binding at mismatched sites (false positives). A two-step cycling protocol with a combined annealing/extension step is also common in diagnostic assays when the annealing temperature is close to the extension temperature.
3. Extension (72°C)
Taq polymerase extends from the 3' end of each annealed primer in the 5' to 3' direction, synthesizing a new DNA strand complementary to the template. The optimal temperature for Taq is 72°C. Extension proceeds at approximately 1000 base pairs per minute.
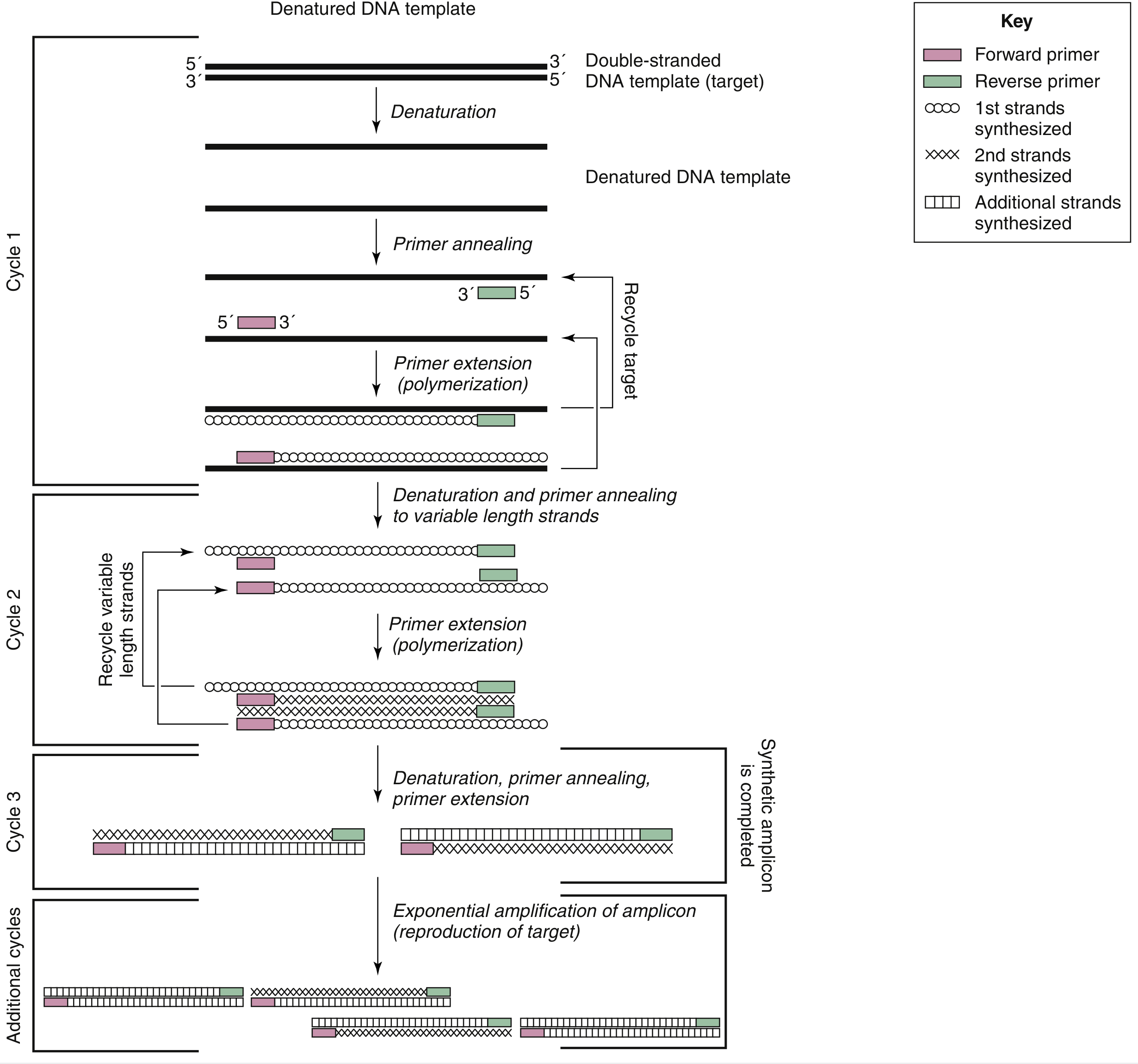
Kinetics of Amplification
After each cycle, the number of copies of the target sequence is theoretically doubled. After n cycles, the target sequence is amplified 2ⁿ-fold. Ideally:
- After 20 cycles: ~1 million-fold amplification
- After 30 cycles: ~1 billion-fold amplification
In practice, amplification is never perfectly efficient due to reaction inhibitors or suboptimal conditions. The more accurate expression is (1 + e)ⁿ, where e is the amplification efficiency (0 < e < 1) and n is the number of cycles. A typical PCR run consists of 25-40 cycles.
Detection of PCR Products (Amplicons)
After amplification, the products (amplicons) are detected by various methods:
- Gel electrophoresis with ethidium bromide staining - The classic approach. Amplicons are separated by size and visualized as bands under UV light. Suitable for most diagnostic applications requiring simple presence/absence results.
- Capillary electrophoresis - Used for finer size discrimination.
- Mass spectrometry - Allows accurate sizing of amplification products.
- Probe hybridization - Southern blotting or dot-blot hybridization with labeled probes to verify amplicon identity.
- Real-time detection - Addition of fluorescent dyes or probes to the reaction mixture allows optical monitoring during amplification (discussed in detail below).
Variants of PCR
Since its introduction, numerous PCR variants have been developed to increase sensitivity, specificity, or expand the range of targets detectable.
1. Reverse Transcriptase PCR (RT-PCR)
Original PCR was designed for DNA amplification. RT-PCR was developed to amplify RNA targets. In this process, complementary DNA (cDNA) is first synthesized from the RNA template by the enzyme reverse transcriptase (RT), and the resulting cDNA is then amplified by conventional PCR. Originally this used two enzymes (a heat-labile RT plus a thermostable DNA polymerase), but thermostable DNA polymerases from Thermus species that can function as both RT and DNA polymerase have simplified the process into a single-enzyme, one-tube reaction. RT-PCR is essential for detecting RNA viruses (HIV, SARS-CoV-2, hepatitis C), and for measuring gene expression levels.
2. Nested PCR
Nested PCR was developed to increase both sensitivity and specificity. It employs two pairs of primers and two successive rounds of amplification. The first primer pair amplifies a large segment in an initial 15-30 cycles. The product from this first round then serves as the template for a second round using a second, "nested" primer pair that anneals to an internal sequence of the first-round product. This approach verifies the identity of the product and greatly increases sensitivity (due to high total cycle number) and specificity (due to the requirement that both primer pairs bind). The major disadvantage is the risk of carryover contamination during the transfer of first-round products to the second tube.
3. Multiplex PCR
Multiplex PCR uses multiple pairs of primers simultaneously in a single reaction to amplify several different target sequences at once. This is highly valuable for detecting multiple pathogens in a single specimen, or for simultaneously assessing multiple mutations. Each primer pair must be optimized so as not to interfere with the others. Examples include panels for respiratory viruses and sexually transmitted infections.
4. Real-Time PCR (Quantitative PCR, qPCR)
Real-time PCR is arguably the most important advance in PCR technology. It allows simultaneous amplification and detection of product during the exponential phase of the reaction, eliminating the need for post-amplification processing. Fluorescent dyes or probes are incorporated into the reaction, and a fluorescence detector in the thermocycler monitors signal accumulation with each cycle. The cycle at which fluorescence crosses a defined threshold is called the Ct (Cycle Threshold) or Cq (Quantification Cycle) value - this inversely correlates with the initial amount of target nucleic acid in the sample.
Detection chemistries in real-time PCR include:
-
Intercalating dyes (e.g., SYBR Green) - These dyes fluoresce when bound to double-stranded DNA. Simple and inexpensive, but non-specific since they bind to any dsDNA product, including primer-dimers. Specificity is confirmed by melt-curve analysis post-amplification.
-
TaqMan (5' nuclease) probes - A sequence-specific fluorogenic probe is added to the reaction. The probe is labeled with a fluorescent reporter dye at its 5' end and a quencher dye at its 3' end. When intact, the quencher absorbs the reporter signal. During primer extension, Taq polymerase's 5' to 3' exonuclease activity degrades the probe, releasing the reporter from proximity to the quencher, generating fluorescence. Signal is generated only when both primer and probe are bound to the same template, providing high specificity.
-
FRET (Fluorescence Resonance Energy Transfer) probes - Two adjacent hybridization probes. The donor dye on one probe transfers energy to the acceptor dye on the adjacent probe when both are hybridized. The emitted signal is proportional to the amount of PCR product.
-
Molecular beacons - Hairpin-shaped probes with a fluorophore at one end and a quencher at the other. When unbound, the stem-loop structure brings fluorophore and quencher together (no fluorescence). On binding to the target sequence, the hairpin opens, separating fluorophore from quencher and restoring fluorescence.
-
Scorpion probes - Combine a PCR primer with a molecular beacon on the same molecule, offering faster hybridization kinetics.
Real-time qPCR is applied to measure viral loads (HIV, hepatitis B/C, cytomegalovirus), monitor minimal residual disease (e.g., BCR-ABL fusion transcripts in CML), quantify gene expression, and for absolute copy number determination.
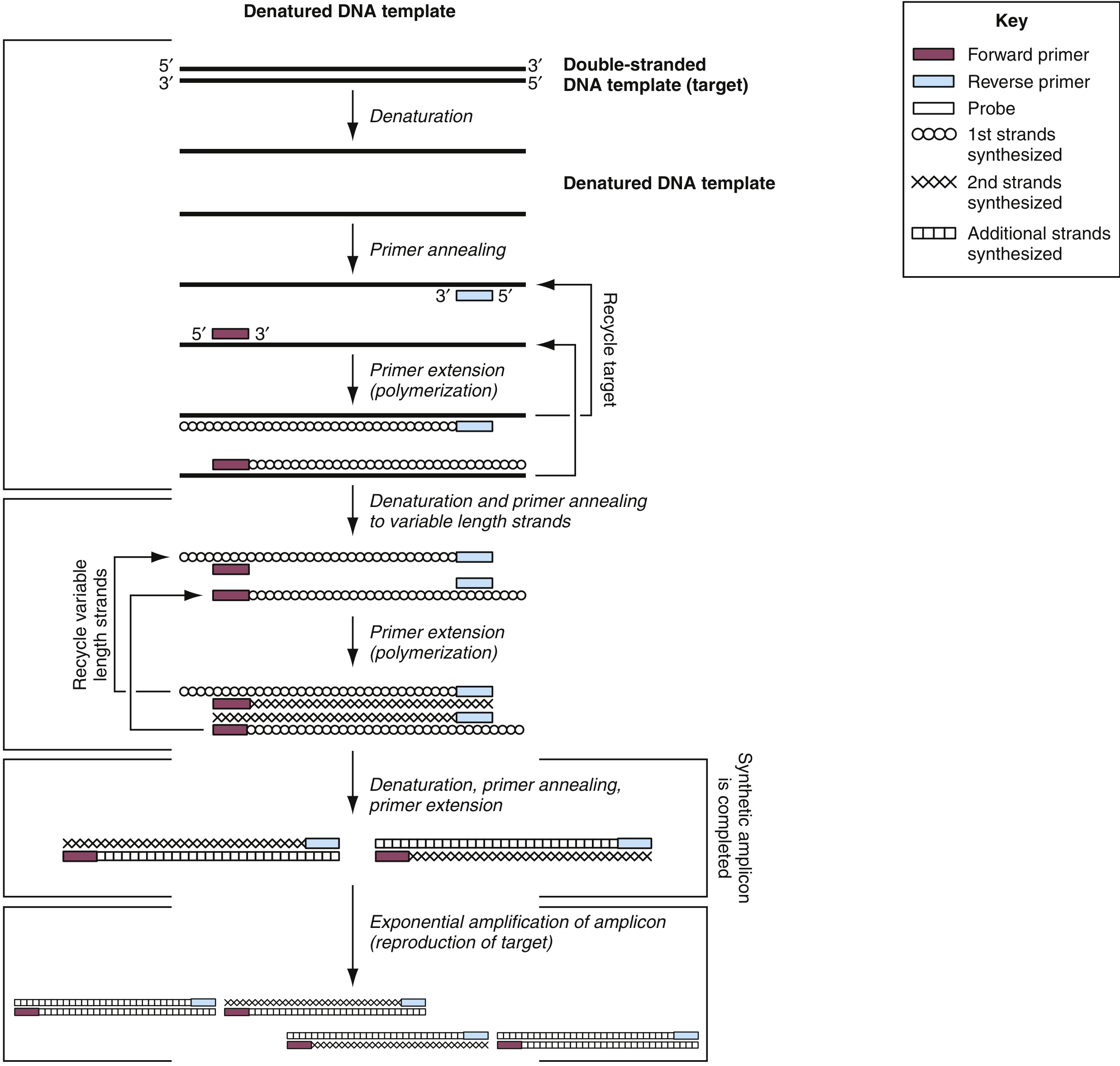
5. Digital PCR (dPCR)
Digital PCR represents the next generation of quantitative PCR. The sample is partitioned into thousands of individual reaction compartments (droplets, micro-wells, or capillaries) before amplification, so each compartment contains either zero or one template molecule. After amplification, compartments are scored as positive or negative, and the original target concentration is calculated using Poisson statistics - no standard curve is required. Digital PCR is particularly suited to:
- Detection and absolute quantification of low-level targets (rare mutations, viral loads)
- Copy number variation analysis
- Rare mutation detection in heterogeneous tumor samples
- Establishing quantitative reference materials
6. Allele-Specific PCR (AS-PCR)
In allele-specific PCR, primers are designed so that the 3' end of a primer matches only one allele at the site of a known mutation or polymorphism. Taq polymerase lacks significant 3' to 5' exonuclease (proofreading) activity, so a mismatch at the 3' end severely impairs extension. Only DNA with the targeted allele is efficiently amplified. Sensitivity can reach 0.1%, making it useful for detecting low-frequency mutations in heterogeneous tumor samples. Examples include detection of IDH1 and TERT promoter mutations in glioma.
7. COLD-PCR (CO-amplification at Lower Denaturation Temperature PCR)
Exploits the lower melting temperature of mutant heteroduplexes to preferentially amplify mutant sequences from a background of wild-type DNA, enriching minor allele variants before sequencing.
PCR in Pathological Diagnosis: Applications
A. Infectious Diseases
PCR has transformed the diagnosis of infectious diseases, offering speed, sensitivity, and the ability to detect organisms that cannot be cultured or are present in too small a quantity for conventional methods.
- Bacterial infections: Detection of Mycobacterium tuberculosis directly from sputum (instead of waiting weeks for culture), MRSA (methicillin-resistant Staphylococcus aureus) detection from swabs, Chlamydia trachomatis, Neisseria gonorrhoeae, Bordetella pertussis, H. pylori (though false negatives can occur due to PCR inhibitors in gastric tissue).
- Viral infections: HIV viral load monitoring by RT-PCR (quantifies circulating virus), hepatitis B and C viral load, CMV in transplant recipients, HSV encephalitis (PCR of CSF is the gold standard), SARS-CoV-2 diagnosis by RT-PCR from nasopharyngeal swabs, respiratory virus panels, avian influenza subtyping.
- Fungal and parasitic infections: Pneumocystis jirovecii, malaria species identification.
- An important caveat: PCR detects nucleic acid, not viable organisms. A positive result does not confirm active infection, and false-negatives can arise from inhibitors in the sample.
B. Oncology and Hematopathology
- Chromosomal translocations: The BCR-ABL fusion gene (Philadelphia chromosome, t(9;22)) in CML and ALL is detected and quantified by RT-PCR. Quantitative RT-PCR is the method of choice for monitoring minimal residual disease (MRD) in CML after treatment with imatinib or other TKIs - serial quantification guides clinical decisions.
- Gene mutations: KRAS, EGFR, BRAF (V600E), IDH1/2, NPM1, FLT3 mutations in various carcinomas and leukemias are detected by allele-specific PCR, real-time PCR, or Sanger sequencing following PCR.
- Lymphoma clonality: PCR-based analysis of immunoglobulin heavy chain (IgH) gene rearrangement distinguishes clonal B-cell proliferations (lymphoma) from polyclonal reactive proliferations.
- T-cell receptor (TCR) gene rearrangement: Analogous to IgH analysis, used to establish T-cell clonality.
- Trinucleotide repeat expansions: Fragile X syndrome (FMR1), Huntington's disease, and other repeat expansion disorders are diagnosed by amplicon length analysis.
- Loss of heterozygosity (LOH): PCR-based microsatellite analysis detects allelic loss, relevant in tumor suppressor gene inactivation.
C. Genetic and Inherited Disorders
- Prenatal diagnosis of single-gene disorders (e.g., sickle cell anemia, cystic fibrosis, thalassemia) from chorionic villus samples or amniocytes.
- Alpha-thalassemia diagnosis by PCR genetic analysis.
- Pharmacogenomics: CYP2D6, CYP2C19 genotyping to predict drug metabolism.
D. Tissue Typing and Transplantation
- HLA typing by PCR-sequence-specific primers (PCR-SSP) or PCR-sequence-specific oligonucleotides (PCR-SSO) for transplant matching.
E. Forensic Pathology
- STR (short tandem repeat) analysis by PCR is the gold standard for DNA fingerprinting in forensic identification, paternity testing, and identification of victims.
Pitfalls and Sources of Error
1. Contamination (False Positives)
The exquisite sensitivity of PCR - capable of detecting a single copy of DNA - is also its greatest weakness with respect to false-positive results. Carryover of amplicons from previous reactions is the most common source of contamination. Even minute aerosol droplets deposited on bench surfaces, pipettes, or reagents can contaminate subsequent reactions. Prevention strategies include:
- Physically separating pre- and post-amplification areas.
- Use of closed-tube (real-time PCR) methods that never expose amplicons to the environment.
- Uracil-DNA glycosylase (UNG) treatment: substituting dUTP for dTTP in PCR reactions, then treating subsequent reactions with UNG before amplification. UNG degrades any uracil-containing carryover amplicons, but not the original genomic DNA (which contains thymine, not uracil).
- Stringent use of dedicated equipment, disposable plasticware, and gloves.
- Negative controls in every run.
2. Inhibitors (False Negatives)
Many substances in clinical specimens can inhibit Taq polymerase and cause false-negative results. These include:
- Hemoglobin and heme compounds (blood samples)
- Bile salts and complex polysaccharides (stool samples)
- Heparin (anticoagulant in blood tubes)
- H. pylori inhibitors in gastric tissue
- Melanin (tissue biopsies)
- Calcium alginate from swabs
Internal controls (added to every sample before extraction) are used to detect inhibition.
3. Primer Design Issues
- Non-specific amplification from primer binding to non-target sequences.
- Primer-dimer formation (primers anneal to each other).
- Hairpin structures within primers.
- These are mitigated by careful bioinformatics-based primer design and optimization of annealing temperature.
4. DNA Degradation
Formalin-fixed, paraffin-embedded (FFPE) tissues present a particular challenge. Formalin crosslinks and fragments DNA, limiting amplification to short fragments (<200-300 bp). Extracted DNA from FFPE requires careful assessment of quality and the use of short amplicons in PCR design.
Quality Assurance
Rigorous quality control is mandatory in clinical PCR laboratories:
- Positive controls - Known positive sample included in each run to confirm the assay worked.
- Negative controls - No-template controls (NTC) to detect contamination.
- Internal controls - Added to every patient sample to detect inhibition and extraction failure.
- MIQE guidelines (Minimum Information for Publication of Quantitative Real-time PCR Experiments) provide standardized reporting requirements for real-time PCR assays.
- Laboratory accreditation and participation in external quality assurance (EQA) schemes.
Microfluidic and Point-of-Care PCR
Microfluidic PCR miniaturizes the PCR reaction onto a chip, reducing volumes and dramatically decreasing thermocycling time. Amplification of 500-997 bp fragments has been achieved in as little as 1.7-3.2 minutes, limited only by the kinetics of nucleotide incorporation by Taq polymerase. Fully integrated microfluidic devices can perform DNA extraction, amplification, and electrophoretic separation in a single closed system. For example, the identification of Bacillus anthracis has been accomplished in only 24 minutes using such a device. Point-of-care PCR platforms (e.g., GeneXpert system) enable rapid molecular diagnostics at or near the patient's bedside, with the turnaround times measured in minutes rather than days.
Comparison with Other Nucleic Acid Amplification Techniques (NAATs)
PCR is not the only nucleic acid amplification technique. It is instructive to compare it with isothermal methods that do not require a thermocycler:
| Feature | PCR | RT-PCR | TMA/NASBA | SDA |
|---|---|---|---|---|
| Target | DNA | RNA | RNA (primarily) | DNA/RNA |
| Temperature cycling | Yes | Yes | No (isothermal) | No (isothermal) |
| Key enzyme | Taq polymerase | RT + Taq | RT + RNA polymerase | Restriction enzyme + polymerase |
| Product | DNA amplicon | DNA amplicon | RNA copies | DNA amplicon |
Transcription-Mediated Amplification (TMA) is isothermal and particularly suited to RNA targets. Strand Displacement Amplification (SDA) uses restriction endonuclease nicking to drive isothermal amplification of both DNA and RNA. These techniques are used in automated clinical platforms for STI testing (chlamydia, gonorrhea).
Future Directions
PCR forms the backbone of next-generation sequencing (NGS) workflows, where target enrichment libraries are built by PCR amplification. Digital PCR is increasingly used alongside NGS for quantification and rare variant detection. Loop-mediated isothermal amplification (LAMP) and CRISPR-Cas-based detection (e.g., SHERLOCK, DETECTR) represent emerging alternatives that may complement or supplement PCR-based diagnostics in resource-limited settings.
Conclusion
PCR has fundamentally reshaped pathological diagnosis since its introduction four decades ago. Its ability to detect and amplify infinitesimal quantities of nucleic acid with extraordinary precision makes it applicable across virtually every subspecialty of pathology - from the bedside diagnosis of infectious disease to the molecular stratification of cancer and the prenatal diagnosis of genetic disorders. Variants such as RT-PCR, nested PCR, multiplex PCR, real-time qPCR, and digital PCR have progressively expanded its utility. The rigorous understanding of its principles, applications, and pitfalls is central to the practice of modern pathology.
Key References:
- Robbins, Cotran & Kumar: Pathologic Basis of Disease, pp. on PCR Detection of DNA and RNA Sequence Alterations
- Henry's Clinical Diagnosis and Management by Laboratory Methods (Saiki et al., 1985; 1988) - Section 69: Polymerase Chain Reaction
- Medical Microbiology, 9e - Chapter 5: Nucleic Acid Amplification Techniques
- Tietz Textbook of Laboratory Medicine, 7th Edition - Molecular Diagnostics Section
This is a shared conversation. Sign in to Orris to start your own chat.