Cockayne syndrome
Reading File
Finding Sources
Searching PubMed
Cockayne syndrome
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive information. Let me compile the full answer.
Cockayne Syndrome
Cockayne syndrome (CS) is a rare autosomal recessive multisystem disorder caused by a defect in transcription-coupled nucleotide excision repair (TC-NER). It was first described by E.A. Cockayne in 1936, characterised by cachectic dwarfism, deafness, and pigmentary retinal degeneration.
Genetics and Molecular Basis
CS results from mutations in two main genes:
| Gene | Alias | Frequency |
|---|---|---|
| CSB | ERCC6 | ~2/3 of cases |
| CSA | ERCC8 | ~1/3 of cases |
Both genes are part of the nucleotide excision repair (NER) pathway. Their specific role is in transcription-coupled repair - recognising and repairing DNA lesions (particularly UV-induced pyrimidine dimers) that stall RNA polymerase during active transcription. When RNA polymerase stalls at a DNA lesion, CSB and CSA recruit the repair machinery and chromatin remodellers to clear the blockage. Without functional TC-NER, transcription is persistently blocked at DNA damage sites, leading to cell death and progressive tissue degeneration.
Additional CS-associated mutations occur in XPB, XPD, and XPG - these produce the XP-Cockayne overlap syndrome, which carries elevated skin cancer risk unlike classical CS.
The inheritance is autosomal recessive with a 1-in-4 recurrence risk for siblings.
- Fitzpatrick's Dermatology, p. 2384-2385
- Andrews' Diseases of the Skin, p. 668
Clinical Subtypes
| Type | Onset | Severity |
|---|---|---|
| CS Type I (classic) | 2nd year of life | Moderate, slowly progressive |
| CS Type II (severe) | Infancy/congenital | Severe, rapid deterioration |
| CS Type III (mild) | Late onset | Mild, longer survival |
| COFS syndrome | Congenital | Most severe (microcephaly, cataracts, arthrogryposis) |
| UVSS | - | Mildest - UV sensitivity only, no systemic features |
Clinical Features
Cutaneous
- Photosensitivity: acute, painful burning on minimal sun exposure (in ~75% of patients) - without the freckling or pigmentary abnormalities of xeroderma pigmentosum
- Photodermatitis with telangiectasia, atrophy, and scarring
- No increase in skin cancer (distinguishes CS from XP)
- Loss of subcutaneous fat giving a "wizened," cachectic appearance
- Nail dystrophy, hair abnormalities, cyanotic acral oedema of hands and feet
- Large, cyanotic-appearing hands and feet
Neurological
- Developmental delay and progressive intellectual deterioration
- Microcephaly, progressive growth failure
- Peripheral neuropathy (primary segmental demyelination), reduced nerve conduction velocities
- Spastic weakness and ataxia; occasionally athetosis
- Sensorineural hearing loss (nerve deafness), progressive
- Normal pressure hydrocephalus
- Tigroid leukodystrophy (segmental myelin loss)
- Basal ganglia calcification (putamen)
- Cerebellar cortical atrophy on pathology
- CSF is normal; no diagnostic biochemical findings
Ocular
- Progressive pigmentary retinal degeneration ("salt-and-pepper" retina), leading to blindness
- Cataracts (early-onset)
- Microphthalmia; sunken eyes (enophthalmia) due to loss of orbital fat
- Photophobia, diminished tearing, conjunctivitis, corneal scarring
- Iris atrophy, poorly reactive pupils
- Pendular nystagmus
- Abnormal electroretinogram
Somatic / Other
-
Stunted growth - evident by 2nd-3rd year
-
Prognathism, beaked/prominent nose, sunken eyes - facies resembling progeria or "bird-headed dwarfism"
-
Anhidrosis, poor lacrimation
-
Dental: tooth enamel hypoplasia
-
Kyphoscoliosis, flexion contractures
-
Cryptorchidism
-
Shortened lifespan
-
Adams and Victor's Principles of Neurology, p. 982
-
Fitzpatrick's Dermatology, p. 2384-2385
Pathology
- Small brain
- Striatal and cerebellar calcifications
- Leukodystrophy resembling Pelizaeus-Merzbacher disease
- Severe cerebellar cortical atrophy
- Peripheral nerves: primary segmental demyelination
Diagnosis
Mandatory major criteria (all three required):
- Developmental delay
- Progressive growth failure
- Progressive microcephaly
Minor criteria (3 out of 5 required):
- Cutaneous photosensitivity
- Pigmentary retinopathy and/or cataracts
- Progressive sensorineural hearing loss
- Tooth enamel hypoplasia
- Enophthalmia (sunken eyes)
Brain imaging criteria (supportive):
- White matter hypomyelination
- Cerebellar atrophy or hypoplasia
- Bilateral calcifications of the putamen
Laboratory testing:
- Cellular UV hypersensitivity assay - CS cells show impaired recovery of RNA synthesis after UV irradiation (while XP cells show reduced unscheduled DNA synthesis - a useful distinguishing test)
- Molecular: sequencing of CSA (ERCC8) and CSB (ERCC6) genes
- Prenatal diagnosis by linkage analysis in families with a known mutation
Comparison: CS vs. Xeroderma Pigmentosum (XP)
| Feature | Cockayne Syndrome | Xeroderma Pigmentosum |
|---|---|---|
| DNA repair defect | TC-NER (transcription-coupled) | GG-NER (global genome) |
| Skin cancer | No increase | Markedly elevated |
| Freckling/pigmentation | Absent | Prominent |
| Dwarfism | Present | Absent |
| Neurodegeneration | Present | Present (in some) |
| Hearing loss | Present | Absent (usually) |
| Deafness | Yes | No |
XP-Cockayne Overlap Syndrome
Patients with mutations in XPB, XPD, or XPG can display features of both XP and CS - they have the skin cancer risk of XP plus the neurological degeneration of CS. This reflects the dual role of TFIIH complex subunits in both transcription and DNA repair.
Management
There is no curative treatment for CS. Management is supportive:
- Sun protection (clothing, sunscreen, avoidance of UV)
- Ophthalmologic care (cataract surgery, lubricating eye drops for poor lacrimation)
- Hearing aids for sensorineural hearing loss
- Physiotherapy and orthopaedic care for spasticity and contractures
- Nutritional support / gastrostomy for feeding difficulties
- Monitoring and treatment of complications (cataracts, hydrocephalus)
- Note: Valproate is contraindicated in Cockayne syndrome - fatal liver failure has been reported (Harriet Lane Handbook, p. 7302)
- Genetic counselling for families
Key Mnemonics / Summary Points
- CS = Cachectic dwarfism + Sensory deficits (deaf, blind) + Sun sensitivity (no cancer)
- Gene: CSB (ERCC6) > CSA (ERCC8) - both = transcription-Coupled repair
- NO skin cancer (TC-NER defect doesn't leave unrepaired lesions in non-transcribed regions for carcinogenesis the same way GG-NER deficiency does)
- XP-CS overlap: mutations in XPB/XPD/XPG - SKIN CANCER RISK returns
Transcription
Finding Sources
Reading File
Reading File
Finding Sources
Loading Image
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have all the material needed for a thorough answer. Let me compile it.
Transcription
Transcription is the process by which genetic information encoded in DNA is copied into RNA. It is the first step of gene expression, following the central dogma: DNA → RNA → Protein.
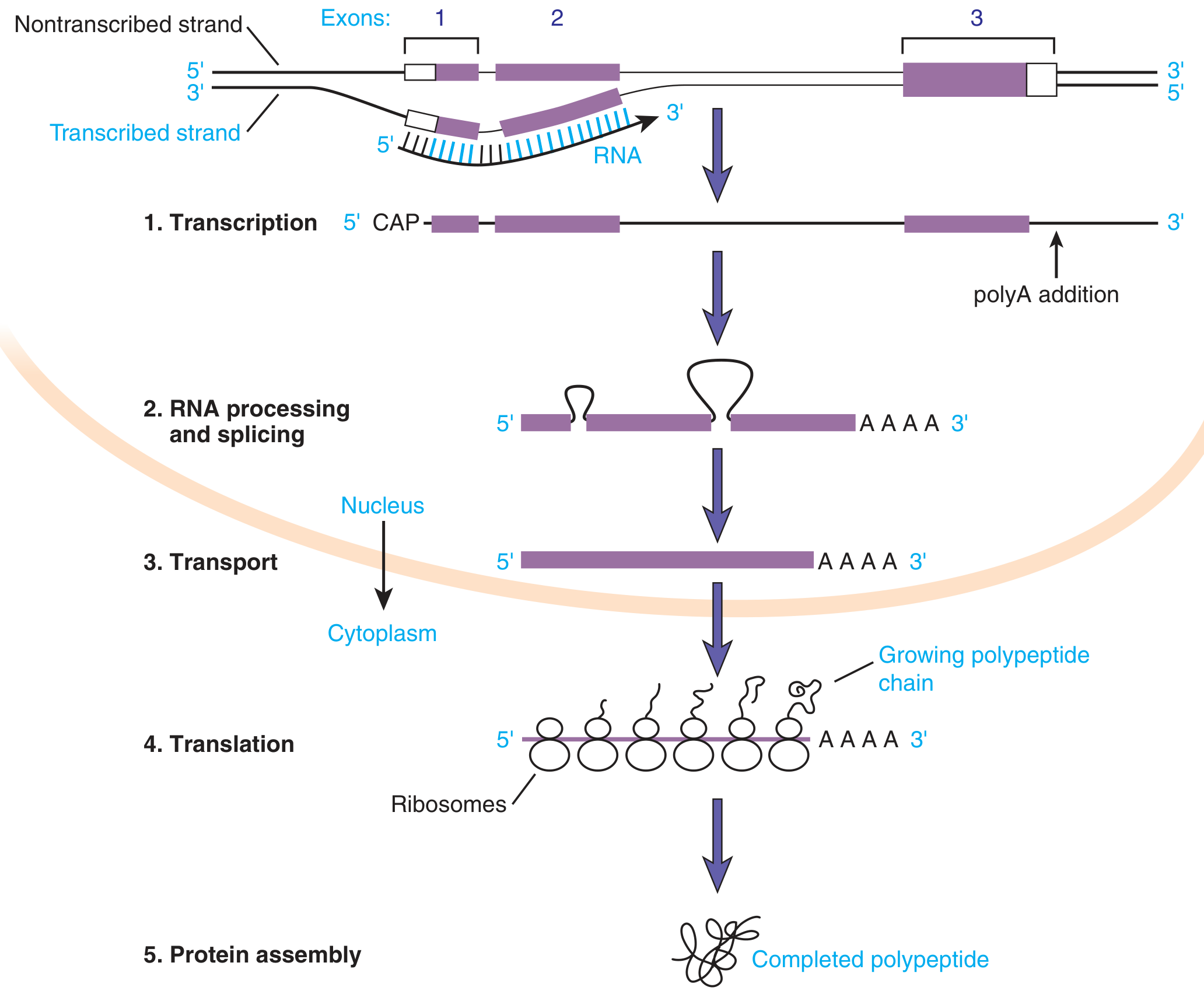
1. Key Terminology: Strands of DNA
| Strand | Also Called | Direction | Role |
|---|---|---|---|
| Template strand | Antisense strand | 3' → 5' | Read by RNA polymerase |
| Coding strand | Sense/non-template strand | 5' → 3' | Identical in sequence to the RNA transcript (with T→U) |
By convention, gene sequences are written as the coding strand, 5' to 3'. The start point of transcription is designated +1; sequences upstream (before it) are negative (-1, -2, etc.).
- Basic Medical Biochemistry, p. 442-444
2. RNA Polymerases
Prokaryotes
A single RNA polymerase (a multi-subunit holoenzyme) handles all transcription. It contains a core enzyme plus a sigma (σ) factor, which confers promoter recognition specificity. The most common is σ70 in E. coli.
Eukaryotes (3 nuclear polymerases)
| Polymerase | Products |
|---|---|
| RNA Pol I | rRNA (28S, 18S, 5.8S) - initially as a 45S primary transcript |
| RNA Pol II | mRNA + microRNA (miRNA) |
| RNA Pol III | tRNA + 5S rRNA + other small RNAs |
The mitochondria has its own separate RNA polymerase for the mitochondrial genome.
-
RNA polymerases, unlike DNA polymerases, do not require a primer to start synthesis.
-
They also lack extensive error-checking (proofreading) capability.
-
Synthesis always proceeds 5' to 3', reading the template 3' to 5'.
-
Basic Medical Biochemistry, p. 442; Tietz Textbook of Laboratory Medicine, p. 1527
3. Promoters
The promoter is a DNA sequence upstream of the transcription start site that specifies where RNA polymerase binds to initiate transcription.
Prokaryotic Promoters (E. coli)
- -10 box (Pribnow box): consensus sequence TATAAT, recognized by sigma factor σ70
- -35 region: consensus sequence TTGACA
Eukaryotic Promoters (RNA Pol II)
-
TATA box (Hogness box): consensus TATA(A/T)A, located ~-25 to -40 from the start site. Found in only ~12.5% of promoters - these are highly regulated genes. Housekeeping genes often lack a TATA box.
-
BRE (TFIIB-recognition element): GC-rich sequence, upstream of TATA
-
Initiator element (Inr): most common core promoter element (~50% of promoters), at the +1 site
-
DPE (downstream promoter element) and MTE (motif ten element): located downstream of +1
-
Promoter-proximal elements: in the -100 to -200 region, bind regulatory proteins
-
Enhancers: distal elements that can be thousands of base pairs upstream or downstream; they stabilize RNA Pol binding but do not define the initiation site. The DNA folds back on itself to allow enhancer-bound transcription factors to interact with the RNA Pol complex.
-
Basic Medical Biochemistry, p. 444
4. Stages of Transcription
Initiation
- Transcription factors bind to the promoter
- For RNA Pol II: TFIID binds the TATA box first, then recruits TFIIA, TFIIB, TFIIE, TFIIF, TFIIH, and RNA Pol II to form the pre-initiation complex (PIC)
- TFIIH (which contains XPB and XPD helicases - relevant to Cockayne syndrome and XP) unwinds the DNA around the start site
- The double-stranded DNA separates; RNA synthesis begins
Elongation
- RNA Pol moves along the template strand 3'→5', synthesizing RNA 5'→3'
- Nucleotides are added to the free 3'-OH of the growing chain
- The DNA strands separate ahead of the polymerase and rejoin behind it (the "transcription bubble")
- No primer is needed
Termination
-
Prokaryotes: a termination factor (Rho) recognizes a DNA termination sequence, OR intrinsic termination occurs via hairpin loop formation in the RNA
-
Eukaryotes (RNA Pol II): termination is coupled to polyadenylation - transcription continues past the coding region until poly(A) signals are encountered, then RNA is cleaved and the polymerase released
-
Tietz Textbook of Laboratory Medicine, p. 1527-1528
5. Post-Transcriptional Processing (Eukaryotes)
The primary transcript (heterogeneous nuclear RNA = hnRNA or pre-mRNA) undergoes three major modifications in the nucleus before it becomes mature mRNA:
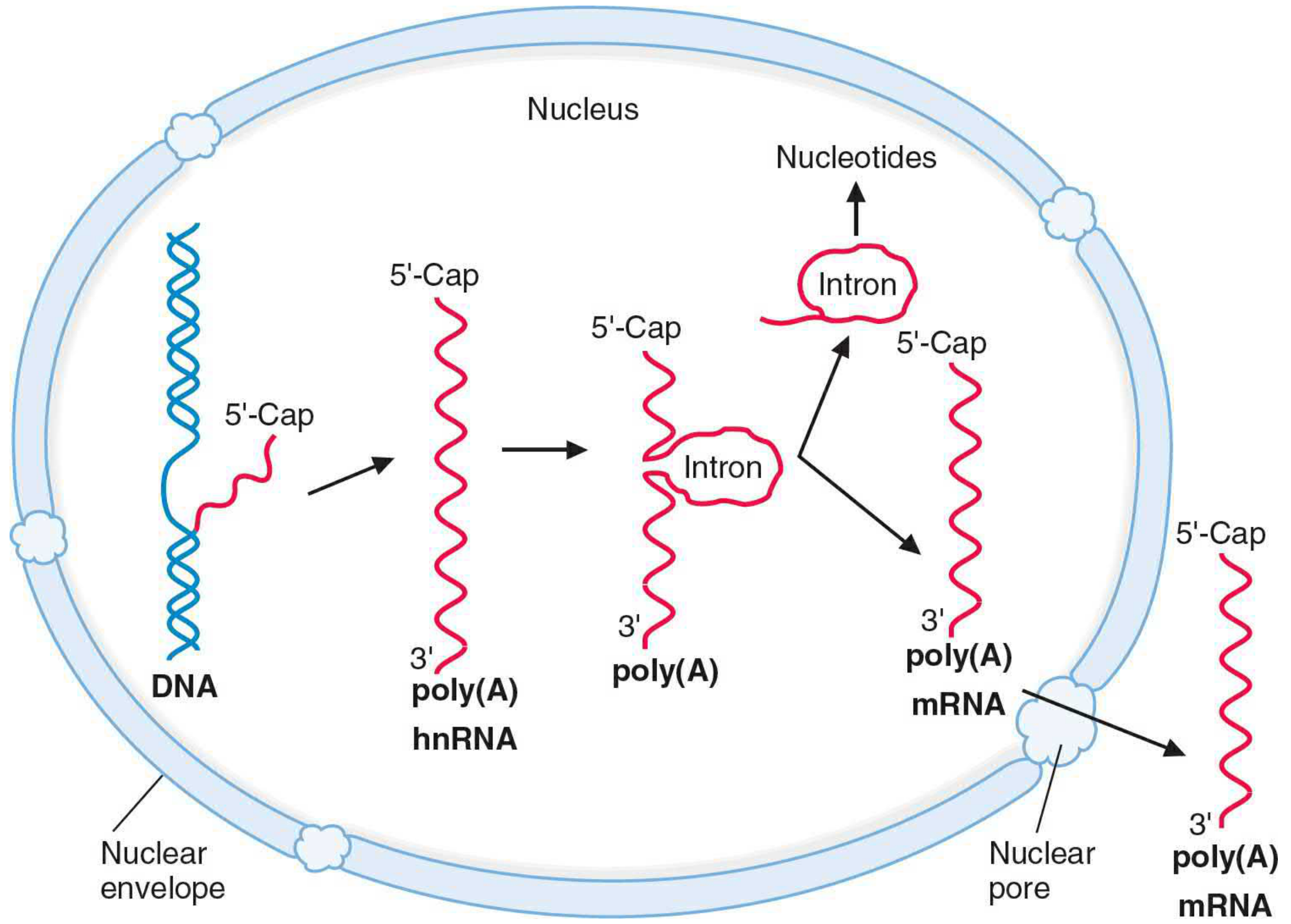
A. 5' Capping
- After ~20-30 nucleotides have been transcribed, a 7-methylguanosine (m7G) cap is added to the 5' end via an unusual 5'→5' triphosphate linkage
- A methyltransferase methylates the N7 position of the G residue
- Three cap types exist: Cap 0, Cap 1, Cap 2 (based on ribose methylation of the first and second nucleotides)
- Functions: protects mRNA from degradation by 5' exonucleases, facilitates nuclear export, and promotes ribosome attachment for translation
B. 3' Polyadenylation
- The signal sequence AAUAAA (or a variant) is encoded in the 3' UTR of the pre-mRNA
- The pre-mRNA is cleaved at a specific point ~10-30 nt downstream of AAUAAA
- Poly(A) polymerase adds ~200 adenylate residues to the 3' end
- Functions: stability of mRNA, nuclear export, and enhancement of translation
- Note: histone mRNAs are a notable exception - they are not polyadenylated
C. RNA Splicing
- Introns are non-coding sequences interspersed between coding exons
- Splicing signals:
- 5' splice site (donor): GT dinucleotide (GU in RNA)
- 3' splice site (acceptor): AG dinucleotide
- Branch site: an intronic sequence ~20-50 nt upstream of the 3' splice site (contains an adenosine residue)
- The spliceosome (a complex of 5 small nuclear RNAs - U1, U2, U4, U5, U6 - and associated proteins called snRNPs / "snurps") catalyzes splicing
- Mechanism: two transesterification reactions form a lariat intermediate (the intron loops back on the branch site A), then exons are joined and the intron lariat is released and degraded
- Alternative splicing allows a single gene to generate multiple different protein isoforms
Clinical relevance: Autoantibodies against snRNPs (Smith antigens) are a hallmark of Systemic Lupus Erythematosus (SLE) - snRNPs were actually discovered using antibodies from SLE patients.
- Emery's Medical Genetics, p. 25-26; Basic Medical Biochemistry, p. 449
6. Other RNA Products
rRNA (RNA Pol I)
- rRNA genes are clustered in the nucleolus
- A single 45S primary transcript is cleaved into 28S, 18S, and 5.8S rRNA
- These combine with ribosomal proteins to form the large (60S) and small (40S) ribosomal subunits
tRNA (RNA Pol III)
- Transcribed as precursor tRNAs, then processed by cleavage and base modification
- Cloverleaf secondary structure with 4 loops:
- D-loop (contains dihydrouridine)
- Anticodon loop (contains the 3-base anticodon)
- TΨC-loop (contains ribothymidine T and pseudouridine Ψ)
- Variable loop
- The 3'-end has the invariant CCA sequence where the amino acid attaches
- RNA Pol III recognizes a split internal promoter within the tRNA gene itself (+8 to +19 and ~30-60 bp downstream)
7. Prokaryote vs. Eukaryote Transcription: Key Differences
| Feature | Prokaryotes | Eukaryotes |
|---|---|---|
| RNA polymerases | 1 | 3 (nuclear) + mitochondrial |
| Location | Cytoplasm | Nucleus |
| Coupled transcription-translation | Yes | No (nuclear membrane separates the processes) |
| Pre-mRNA processing | Minimal | Extensive (capping, splicing, polyadenylation) |
| Introns | Rare | Present in most genes |
| One mRNA = one protein? | No (polycistronic) | Yes (monocistronic) |
| Promoter | -10 and -35 boxes | TATA box, Inr, BRE, DPE, MTE |
8. Regulation of Transcription
Transcription is the primary level at which gene expression is regulated:
- Transcription factors: proteins that bind specific DNA sequences (via helix-turn-helix, zinc finger, leucine zipper domains) and either activate or repress RNA Pol binding
- Activators: bind enhancers or promoter-proximal elements; stabilize RNA Pol at the promoter; increase transcription rate
- Repressors: bind the promoter or operator region and block RNA Pol binding, reducing or silencing transcription
- Transcription factors generally bind as dimers and have at least 3 functional domains: DNA-binding, dimerization, and transcriptional activation/repression
- Additional regulation by epigenetic mechanisms (DNA methylation, histone acetylation/methylation) that control chromatin accessibility
9. Clinically Relevant Inhibitors
| Drug/Toxin | Mechanism | Relevance |
|---|---|---|
| Rifampin | Inhibits bacterial RNA polymerase (binds β subunit) | Treatment of tuberculosis |
| α-Amanitin (death cap mushroom) | Inhibits eukaryotic RNA Pol II (and at high doses, Pol III) | Mushroom poisoning; no antidote |
| Actinomycin D | Intercalates DNA, blocks elongation by all RNA polymerases | Used in cancer research/chemotherapy |
- Basic Medical Biochemistry, p. 465
Summary Flow
DNA (double-stranded)
↓ [RNA Polymerase + Transcription Factors bind promoter]
Primary RNA Transcript (pre-mRNA / hnRNA)
↓ [5' Capping]
↓ [3' Polyadenylation]
↓ [Splicing — spliceosome removes introns]
Mature mRNA
↓ [Nuclear export through nuclear pores]
Cytoplasm → Ribosomes → TRANSLATION → Protein
Post translational modifications
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Loading Image
Finding Sources
I have comprehensive data from multiple textbooks. Let me now compile the full answer.
Post-Translational Modifications (PTMs)
Post-translational modification (PTM) is any covalent chemical change to a protein that occurs after translation is complete. PTMs adjust a protein's physical and chemical properties, conformation, stability, activity, subcellular localization, and interactions - massively expanding the functional diversity of the proteome far beyond what the ~20,000 human genes alone could encode.
The most common naturally occurring PTMs are: acetylation, phosphorylation, ubiquitination, glycosylation, methylation, and proteolytic cleavage.
- Rheumatology (Elsevier 2022), p. 2240
- Emery's Elements of Medical Genetics, p. 26
1. Phosphorylation
What: Addition of a phosphate group (–PO₄²⁻) to the hydroxyl group of serine, threonine, or tyrosine residues.
Enzymes: Protein kinases add phosphate (using ATP); protein phosphatases remove it - making phosphorylation rapidly reversible.
Effects:
- Activates or inactivates enzymes
- Creates docking sites for other signaling proteins (e.g., SH2 domains recognize phosphotyrosine)
- Drives conformational changes
Key roles:
- Central mechanism of intracellular signal transduction (e.g., receptor tyrosine kinases, MAPK cascade, JAK-STAT)
- Regulation of cell cycle (cyclin-dependent kinases)
- Metabolic regulation (glycogen phosphorylase, pyruvate dehydrogenase)
Clinical relevance: Dysregulated kinases drive cancer (e.g., BCR-ABL in CML → imatinib target). Phosphatases like SHP-1 shut down immune signaling - their loss causes autoimmunity.
2. Glycosylation
Addition of sugar (carbohydrate) chains to proteins. Two main types:
N-linked glycosylation
- Sugars are attached to the amide nitrogen of asparagine (Asn) in the sequon Asn-X-Ser/Thr (X ≠ Pro)
- Begins in the rough ER: a 14-sugar oligosaccharide (GlcNAc + mannose + glucose) pre-assembled on dolichol phosphate is transferred en bloc to the Asn
- Further processed/trimmed in ER and Golgi - glucose and some mannose residues are removed, then other sugars (GlcNAc, galactose, fucose, NANA/sialic acid) are added
- Tunicamycin blocks N-linked glycosylation
- Congenital Disorders of Glycosylation (CDG): rare syndromes from defects in N-linked glycosylation
- Mannose-6-phosphate (M6P) targeting: in the Golgi, N-linked glycoproteins destined for lysosomes are phosphorylated at mannose residues; M6P receptors recognize this tag and direct vesicles to lysosomes
- I-Cell Disease: GlcNAc phosphotransferase deficiency → no M6P tag → lysosomal enzymes secreted extracellularly instead of reaching lysosomes
O-linked glycosylation
- Sugars attached to the hydroxyl of serine or threonine
- Assembled one sugar at a time in the Golgi (not en bloc)
- GalNAc is the first sugar added (O-GalNAc glycans, "mucin-type")
- Less complex consensus sequence requirement
Functions of glycosylation: protein stability, protection from proteolysis, cell-cell recognition, immune function (ABO blood groups), antigenicity, receptor binding.
- Medical Physiology, p. 899-903
- Biochemistry (Lippincott 8e), p. 482-484
3. Ubiquitination
What: Covalent attachment of the small protein ubiquitin (8.5 kDa, 76 amino acids) to lysine residues of target proteins.
Enzymatic cascade (three steps, requires ATP):
| Enzyme | Name | Function |
|---|---|---|
| E1 | Ubiquitin-activating enzyme | Activates ubiquitin in ATP-dependent reaction |
| E2 | Ubiquitin-conjugating enzyme | Transfers ubiquitin from E1 to E3 |
| E3 | Ubiquitin ligase | Selects the substrate; transfers ubiquitin to target protein's Lys |
A polyubiquitin chain (≥4 ubiquitins) is required to target a protein for proteasomal degradation.
Two key linkage types - linked through different lysines of ubiquitin itself:
| Linkage | Effect |
|---|---|
| K48-linked polyubiquitin | Targets protein to 26S proteasome for degradation |
| K63-linked polyubiquitin | Acts as signaling scaffold (e.g., NF-κB activation via TRAF6) - NOT degradation |
Single ubiquitin or di-ubiquitin on membrane receptors targets them to lysosomes for degradation.
The 26S Proteasome
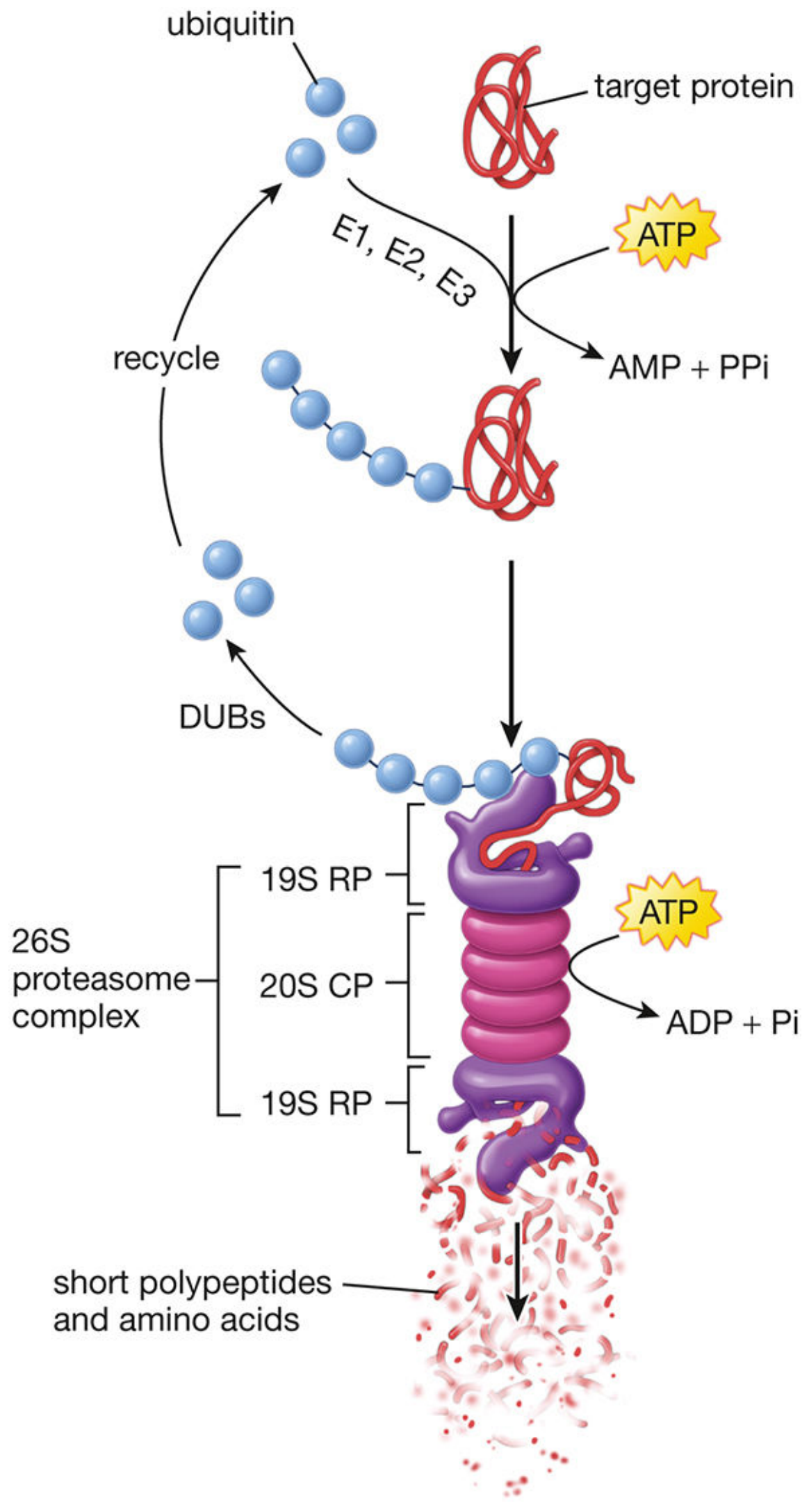
- 20S core particle (CP): barrel-shaped, contains proteolytic active sites - cleaves polyubiquitinated protein into short peptides and amino acids
- 19S regulatory particles (RP): caps on both ends - the "lid" recognizes polyubiquitin tags and unfolds the protein; the "base" releases degradation products
- DUBs (deubiquitinating enzymes): remove and recycle ubiquitin
Targets of ubiquitin-proteasome degradation:
- Misfolded or denatured proteins
- Short-lived regulatory proteins: mitotic cyclins, transcription factors, tumor suppressors/promoters
- Proteins tagged by E3 ligases after phosphorylation (e.g., Cbl ligase targets phosphotyrosine proteins)
Clinical relevance:
-
Proteasome inhibitors (e.g., bortezomib) are used to treat multiple myeloma
-
Parkinson's disease: α-synuclein aggregates partly due to impaired ubiquitin-proteasome function
-
Cancer: many oncoproteins escape ubiquitin-mediated degradation
-
Histology (Gartner), p. 169-170
-
Janeway's Immunobiology 10e, p. 286-287
4. Acetylation
What: Addition of an acetyl group (–COCH₃) from acetyl-CoA to the ε-amino group of lysine (or the α-amino terminus).
Histone acetylation (most studied):
- Catalyzed by HATs (histone acetyltransferases) - opens chromatin for transcription
- Reversed by HDACs (histone deacetylases) - closes chromatin, represses transcription
- Mechanism: acetylation neutralizes the positive charge of lysine, reducing its attraction to the negatively charged DNA backbone → chromatin relaxes → transcription factors gain access
Non-histone proteins: acetylation can regulate protein stability (e.g., acetylation of p53 activates it), protein-protein interactions, and enzyme activity.
Clinical relevance:
-
HDAC inhibitors (e.g., vorinostat) are used as cancer therapies
-
HAT/HDAC imbalance contributes to cancer, inflammatory diseases (rheumatoid arthritis, asthma), and neurodegeneration
-
Rheumatology, p. 3692-3714
5. Methylation
What: Addition of a methyl group (–CH₃) to lysine or arginine residues by methyltransferases, using S-adenosylmethionine (SAM) as the methyl donor. Removed by demethylases.
Unlike acetylation, methylation does NOT change the charge of lysine - its effects depend entirely on which residue is methylated and by how many methyl groups.
Histone methylation and gene regulation (context-dependent):
| Modification | Association |
|---|---|
| H3K4me, H3K36me, H3K79me | Active transcription |
| H3K9me, H3K27me | Repressed transcription / heterochromatin |
This differential pattern constitutes part of the "histone code" - the idea that combinations of histone modifications specify transcriptional outcomes.
DNA methylation (related but distinct PTM of the DNA itself):
-
Methylation of cytosine at CpG dinucleotides → recruits methyl-CpG-binding proteins (MeCP2, MBD1-3) → tethers HDAC complexes → chromatin silencing
-
Critical for genomic imprinting (e.g., Prader-Willi, Angelman, Beckwith-Wiedemann syndromes)
-
Medical Physiology, p. 147
-
Harrison's Internal Medicine 22e
6. Proteolytic Cleavage
What: Irreversible removal of part of the polypeptide chain by protease cleavage.
Types and examples:
| Type | Example |
|---|---|
| Signal peptide removal | Cleavage of N-terminal signal sequence in ER lumen immediately after translocation |
| Zymogen activation | Trypsinogen → trypsin; pepsinogen → pepsin; prothrombin → thrombin |
| Prohormone processing | Proinsulin → insulin + C-peptide; pro-opiomelanocortin → ACTH, β-endorphin, MSH |
| Viral polyprotein cleavage | HIV-1 protease cleaves the gag-pol polyprotein (target of protease inhibitors) |
| Caspase activation | Apoptotic cascade - procaspases cleaved to active caspases |
- Emery's Medical Genetics, p. 26
7. Disulfide Bond Formation
What: Covalent oxidation between two cysteine thiol (–SH) groups to form –S–S–.
Where: Only in oxidizing compartments - the ER lumen and extracellular space. The cytosol is a reducing environment and does not support disulfide bonds.
Enzyme: Protein disulfide isomerase (PDI) in the ER lumen - catalyzes both formation and rearrangement of disulfide bonds until the thermodynamically most stable conformation is reached.
Importance: Stabilizes tertiary and quaternary protein structure (e.g., immunoglobulins, collagen, insulin - the A and B chains of insulin are held together by disulfide bonds).
8. Hydroxylation
What: Addition of –OH to proline → hydroxyproline, and lysine → hydroxylysine.
Key example - Collagen synthesis:
- Prolyl and lysyl hydroxylases act on collagen chains in the ER
- Requires Vitamin C (ascorbate) as an essential cofactor
- Hydroxyproline stabilizes the collagen triple helix through hydrogen bonding
- Hydroxylysine provides sites for glycosylation and for crosslinking between collagen fibrils
Scurvy: Vitamin C deficiency → failure of proline hydroxylation → unstable collagen → bleeding gums, poor wound healing, perifollicular hemorrhages.
- Basic Medical Biochemistry, p. 1887
9. Lipid Modifications (Membrane Targeting)
These modifications anchor proteins to cell membranes:
| Modification | Lipid | Location | Example |
|---|---|---|---|
| Myristoylation | Myristic acid (C14) | N-terminus (Gly) | Src kinase |
| Palmitoylation | Palmitic acid (C16) | Cys residues | Ras, G proteins |
| Prenylation (farnesylation, geranylgeranylation) | Isoprenoid | C-terminus (CAAX motif) | Ras, Rho GTPases |
| GPI anchor | Glycosylphosphatidylinositol | C-terminus | CD55, CD59, prion protein |
GPI-linked proteins: synthesized as transmembrane proteins in the ER, then their lumen-facing domain is cleaved and transferred to the GPI lipid anchor. They remain in the outer leaflet of the plasma membrane.
Paroxysmal nocturnal hemoglobinuria (PNH): defect in GPI anchor synthesis (PIG-A gene mutation) → loss of CD55 and CD59 from RBC surfaces → uncontrolled complement activation → hemolysis.
- Medical Physiology, p. 907
10. Other Important PTMs
| PTM | Target residue | Key function |
|---|---|---|
| SUMOylation | Lys | Protein-protein interactions, nuclear transport, transcription regulation |
| Nitrosylation | Cys (S-nitrosylation) | Signaling by nitric oxide |
| Carboxylation | Glu → γ-carboxyglutamate | Clotting factors II, VII, IX, X require Vitamin K; bind Ca²⁺ |
| ADP-ribosylation | Arg, Glu | Bacterial toxins (cholera, diphtheria); DNA repair (PARP) |
| Formylation | Various | Detected by MS; changes protein weight |
Summary Table of Major PTMs
| PTM | Residue Targeted | Reversible? | Primary Functions |
|---|---|---|---|
| Phosphorylation | Ser, Thr, Tyr | Yes (phosphatases) | Signaling, enzyme regulation |
| N-linked glycosylation | Asn | Partially | Folding, stability, trafficking, immunity |
| O-linked glycosylation | Ser, Thr | Yes | Mucin structure, signaling |
| Ubiquitination (K48) | Lys | Yes (DUBs) | Proteasomal degradation |
| Ubiquitination (K63) | Lys | Yes (DUBs) | Signaling scaffold |
| Acetylation | Lys, N-terminus | Yes (HDACs) | Chromatin opening, protein stability |
| Methylation | Lys, Arg | Yes (demethylases) | Chromatin code, activation/repression |
| Proteolytic cleavage | Peptide bond | No | Activation of zymogens/proproteins |
| Disulfide bond | Cys | Yes (PDI/reductases) | Structural stability |
| Hydroxylation | Pro, Lys | No | Collagen structure |
| Lipidation | Gly/Cys/CAAX | Partial | Membrane anchoring |
This is a shared conversation. Sign in to Orris to start your own chat.