Types of hypersensitivity reaction
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive content from multiple authoritative sources. Let me compile the full answer.
Types of Hypersensitivity Reactions (Gell and Coombs Classification)
Hypersensitivity is defined as an exaggerated or augmented immune response that is harmful to the host. It requires a presensitized state - reactions typically occur after the second encounter with a specific antigen (allergen). In 1963, Coombs and Gell classified hypersensitivity into four types (I-IV), later modified by Janeway et al. (2001). A fifth type and an innate category have since been added.
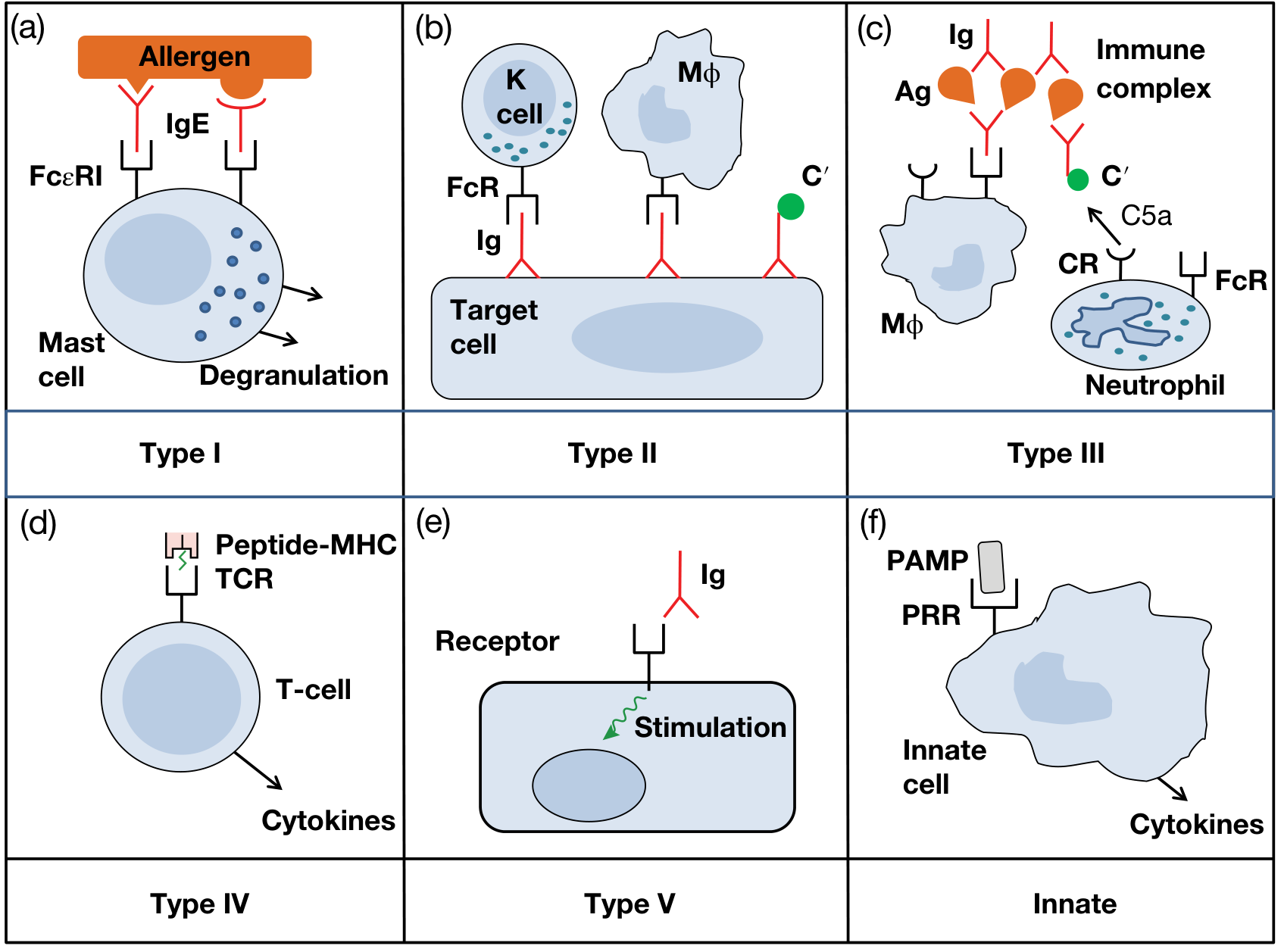
Type I - Immediate (IgE-Mediated) Hypersensitivity
| Feature | Detail |
|---|---|
| Mediator | IgE antibody |
| Cells involved | Mast cells, basophils, eosinophils |
| Onset | Within seconds to minutes of re-exposure |
| Mechanism | Antigen cross-links cell-bound IgE on mast cells → degranulation → release of pharmacologically active mediators |
Mediators released:
- Primary (preformed): Histamine - causes vasodilation, increased capillary permeability, bronchospasm
- Secondary (newly formed): Prostaglandins and leukotrienes (derived from arachidonic acid): LTB4 (chemoattractant), LTC4 and LTD4 (vasodilation, vascular permeability), TNF-alpha, IL-4
Clinical examples:
- Systemic anaphylaxis (e.g., after penicillin, bee sting)
- Allergic asthma
- Allergic rhinitis / hay fever
- Urticaria and angioedema
- Atopic eczema, food allergy
Atopy: A subgroup with strong familial predisposition, elevated IgE levels, and classic presentations (hay fever, asthma, eczema).
Treatment: Epinephrine (first-line for anaphylaxis), antihistamines, corticosteroids; avoidance of the allergen.
Type II - Antibody-Dependent Cytotoxic Hypersensitivity
| Feature | Detail |
|---|---|
| Mediator | IgG (or IgM) antibodies |
| Target | Cell surface or extracellular matrix antigens |
| Onset | Hours |
| Mechanism | Antibody binds cell-surface antigen → complement activation → cell lysis via MAC, opsonization for phagocytosis, or ADCC by NK cells |
Clinical examples:
- Hemolytic anemia (autoimmune or drug-induced, e.g., penicillin-coated RBCs)
- ABO blood transfusion reactions
- Rh hemolytic disease of the newborn
- Goodpasture syndrome (anti-GBM antibody - kidney and lung basement membranes)
- Myasthenia gravis (antibodies to acetylcholine receptors - but note: blocking, not stimulating, so some classify as a variant)
Note: When antibodies stimulate rather than destroy (as in Graves' disease), this is now sometimes classified separately as Type V.
Type III - Immune Complex-Mediated Hypersensitivity
| Feature | Detail |
|---|---|
| Mediator | IgG antibodies complexed with soluble antigen |
| Onset | 6-12 hours (Arthus); days-weeks (serum sickness) |
| Mechanism | Persistent antigen-antibody complexes are deposited in tissues → complement activation → neutrophil/macrophage recruitment → inflammation and tissue damage |
Two main forms:
- Arthus reaction (local): Low-dose antigen injected into skin → IgG + complement + mast cell/neutrophil mediators → local necrotic reaction within 12 hours
- Serum sickness (systemic): Large amount of foreign protein/antigen → circulating immune complexes deposited in joints, kidneys, blood vessels
Clinical examples:
- Post-streptococcal glomerulonephritis (lumpy IgG + C3 deposits along GBM on immunofluorescence)
- Serum sickness
- Vasculitis
- Subacute bacterial endocarditis
- Systemic lupus erythematosus (lupus nephritis)
- Rheumatoid arthritis (joint deposition)
- Hypersensitivity pneumonitis (farmer's lung - fungal spore antigens)
Type IV - Cell-Mediated (Delayed-Type) Hypersensitivity
| Feature | Detail |
|---|---|
| Mediator | Sensitized T cells (no antibody involved) |
| Onset | 48-72 hours (hence "delayed") |
| Mechanism | Antigen-presenting cells activate sensitized T cells → T cell proliferation, cytokine release (IFN-gamma, IL-2) → macrophage activation, inflammation, tissue damage |
T cell subtypes involved (Janeway's modification):
| Subtype | Effector | Example |
|---|---|---|
| Th1 | Macrophage activation | Tuberculin reaction, contact dermatitis |
| Th2 | Eosinophil activation | Chronic allergic asthma |
| Th17 | Neutrophil/macrophage activation | Rheumatoid arthritis, atopic dermatitis |
| Cytotoxic T cells | Direct cell killing | Poison ivy, graft rejection, viral-infected cell killing |
Clinical examples:
- Tuberculin (Mantoux) test - classic example
- Contact dermatitis (nickel, poison ivy, poison oak, topical drugs)
- Granulomatous diseases (tuberculosis, leprosy, sarcoidosis)
- Graft rejection
- Some forms of eczema
Type V - Stimulatory Hypersensitivity (added to the original classification)
| Feature | Detail |
|---|---|
| Mediator | IgG antibody acting as a receptor agonist |
| Mechanism | Antibody binds a cell surface receptor and mimics the normal ligand, continuously stimulating the cell (no cytotoxicity) |
Clinical example:
- Graves' disease: Anti-TSH receptor antibody acts as a TSH agonist, constitutively stimulating the thyroid → hyperthyroidism. Unlike TSH, the antibody is not subject to negative feedback, so stimulation is continuous.
- Agonistic autoantibodies to angiotensin II AT1 receptor in preeclampsia and hypertension.
Innate Hypersensitivity (additional category)
Excessive activation of pattern recognition receptors (PRRs) (e.g., TLRs) on innate cells by PAMPs such as LPS leads to massive cytokine release (TNF, IL-1beta, IL-6) - causing toxic shock, ARDS, and DIC, as seen in Gram-negative sepsis. Superantigens from Staphylococcus/Streptococcus directly cross-link T-cell receptors and provoke massive cytokine release (toxic shock syndrome) via a related mechanism.
Quick Comparison Summary
| Type | Name | Antibody/Cell | Onset | Classic Example |
|---|---|---|---|---|
| I | Immediate / Anaphylactic | IgE | Minutes | Anaphylaxis, asthma, hay fever |
| II | Cytotoxic | IgG/IgM vs. cell surface | Hours | Transfusion reaction, Goodpasture |
| III | Immune complex | IgG + soluble Ag | 6-12 h / days | Post-strep GN, serum sickness, SLE |
| IV | Delayed / Cell-mediated | T cells (no Ab) | 48-72 h | Tuberculin test, contact dermatitis |
| V | Stimulatory | IgG agonist Ab | Variable | Graves' disease |
| Innate | - | Innate PRR activation | Hours | Gram-neg sepsis, toxic shock |
Sources: Jawetz, Melnick & Adelberg's Medical Microbiology 28e; Goldman-Cecil Medicine; Roitt's Essential Immunology; Janeway's Immunobiology 10e
Pathogenesis of gas gangrene
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Excellent - I now have comprehensive content from multiple authoritative sources. Let me compile the full detailed answer.
Pathogenesis of Gas Gangrene (Clostridial Myonecrosis)
Causative Organisms
Gas gangrene is caused by histotoxic Clostridium species - anaerobic, Gram-positive, spore-bearing bacilli:
- C. perfringens (type A) - responsible for ~90% of cases; the dominant pathogen
- C. novyi and C. septicum - next most common; C. septicum notably causes spontaneous gas gangrene without an obvious wound (associated with colonic malignancy)
- C. histolyticum, C. bifermentans (less common)
These organisms are widely found in soil and feces. Spores may originate from the patient's own intestinal flora or from the environment.
Predisposing Conditions (Enabling Pathogenesis)
For infection to develop, three conditions must coexist:
- Wound contamination with spores - from soil, clothing, dirt, or bowel flora
- Devitalized (avascular/necrotic) tissue - provides the anaerobic, low redox potential microenvironment essential for spore germination and bacterial multiplication
- Significant delay between injury and surgical management - allowing time for bacterial proliferation and toxin production
Classic settings:
- High-velocity gunshot or shrapnel wounds (wartime) - cavitation sucks in foreign material, leaving devascularized tissue
- Compound fractures, crush injuries
- Limb ischemia (peripheral vascular disease, diabetics)
- Postoperative wounds, especially bowel surgery
- Septic/incomplete abortions (C. perfringens infects retained necrotic products of conception)
- Spontaneous gas gangrene - bacteremia from a GI source (especially C. septicum) seeds muscle, without any external wound
Step-by-Step Pathogenesis
Step 1 - Spore Germination
Spores deposited in devitalized tissue encounter a critically low oxidation-reduction (redox) potential. This anaerobic environment triggers germination of spores into actively metabolizing vegetative bacteria.
Step 2 - Rapid Bacterial Multiplication
Vegetative clostridia multiply rapidly. Their fermentative metabolism produces CO₂ and H₂ gas (from carbohydrate and protein breakdown), which accumulates in the tissues - the hallmark that gives the disease its name. This gas production further lowers tissue oxygen tension, creating a self-perpetuating cycle that favors continued anaerobic growth.
Step 3 - Toxin Production (the key pathogenic step)
C. perfringens produces at least 12 exotoxins, classified by four major toxin types (alpha, beta, epsilon, theta). The most pathogenically important are:
| Toxin | Type | Mechanism | Effect |
|---|---|---|---|
| Alpha-toxin (lecithinase, phospholipase C) | Primary, major | Hydrolyzes phosphatidylcholine in cell membranes; activates arachidonic acid pathway | Destroys muscle cell membranes, lyses RBCs (hemolysis), destroys platelets and PMNs, causes widespread capillary damage, leads to shock |
| Theta-toxin (perfringolysin O) | Pore-forming cytolysin | Inserts into cholesterol-containing membranes, forming pores | Cytolysis of many cell types; contributes to tissue necrosis and vascular damage |
| Collagenase (kappa toxin) | Collagen-splitting | Degrades collagen in connective tissue | Facilitates spread through tissue planes |
| Hyaluronidase (mu toxin) | Spreading factor | Degrades hyaluronic acid | Allows spread through connective tissue matrix |
| Other proteases | Various | Degrade structural proteins | Further tissue destruction |
Alpha-toxin is the master toxin - it is the major cause of both local tissue destruction and the systemic shock seen in gas gangrene. Fatal cases can occur without bacteremia, strongly suggesting that circulating alpha-toxin absorption drives the systemic effects.
Step 4 - Propagation of Necrosis and the "Leukocyte Block"
A cardinal and unique histological feature of gas gangrene is the near-complete absence of inflammatory cells (leukocytes) within necrotic tissue, despite accumulation of leukocytes in adjacent vessels. This occurs because:
- Alpha-toxin and theta-toxin destroy PMNs directly
- Toxins increase vascular permeability, causing massive edema that impairs leukocyte migration
- Platelet aggregation and microvascular occlusion further impair tissue perfusion
This "leukocyte block" means the host cannot mount an effective local inflammatory defense, allowing unimpeded bacterial spread. The infection progresses along muscle bundles at a rate of up to 10 cm per hour.
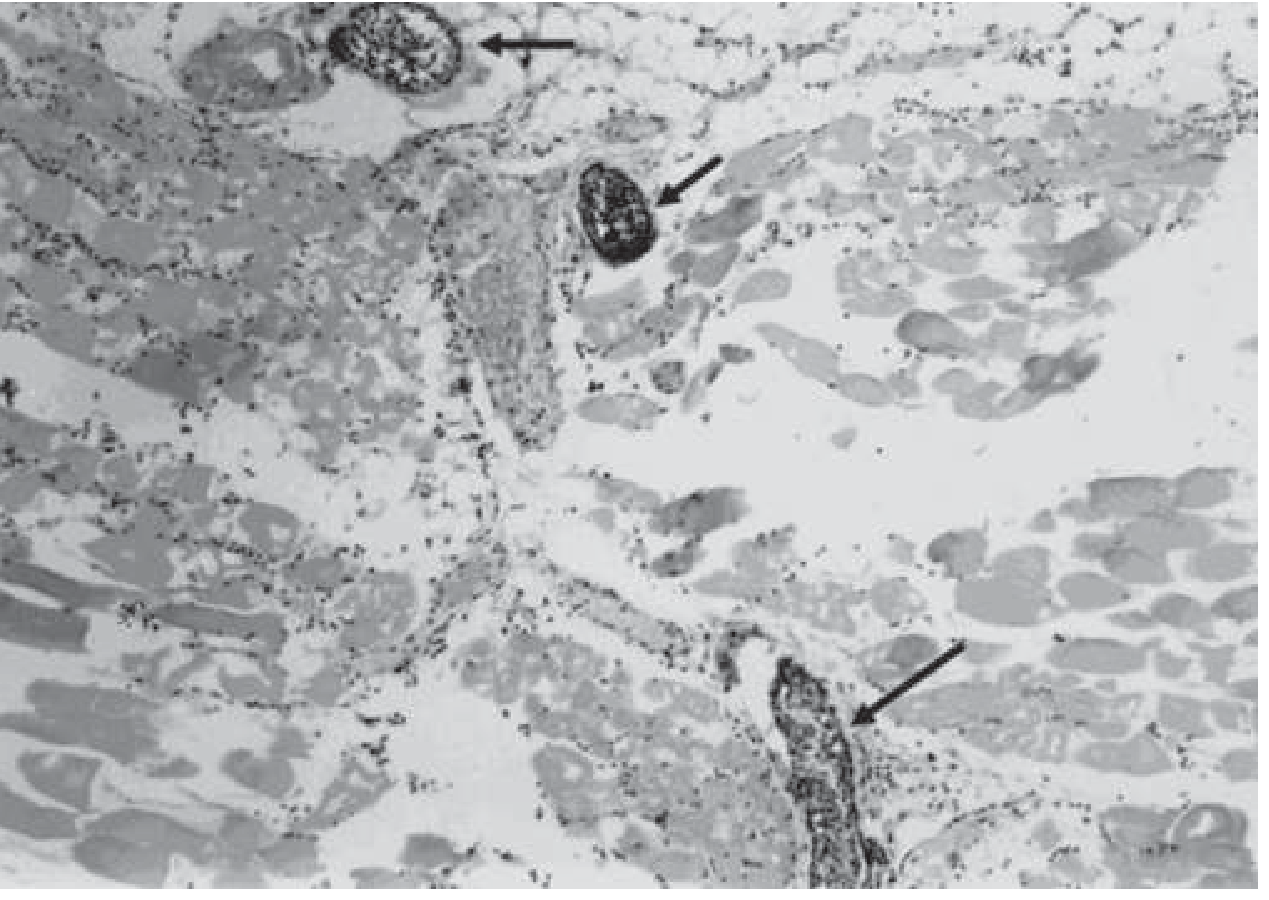
Step 5 - Systemic Effects and Shock
As infection spreads, alpha-toxin is systemically absorbed:
- Intravascular hemolysis - RBC destruction releases hemoglobin → jaundice, hemoglobinuria
- Increased vascular permeability → massive edema and fluid sequestration
- Hypotension and circulatory collapse (shock)
- Renal failure (from hemoglobinuria + hypoperfusion)
- Coma and death - often within 48 hours if untreated
Remarkably, patients remain alert and lucid until very late stages of shock.
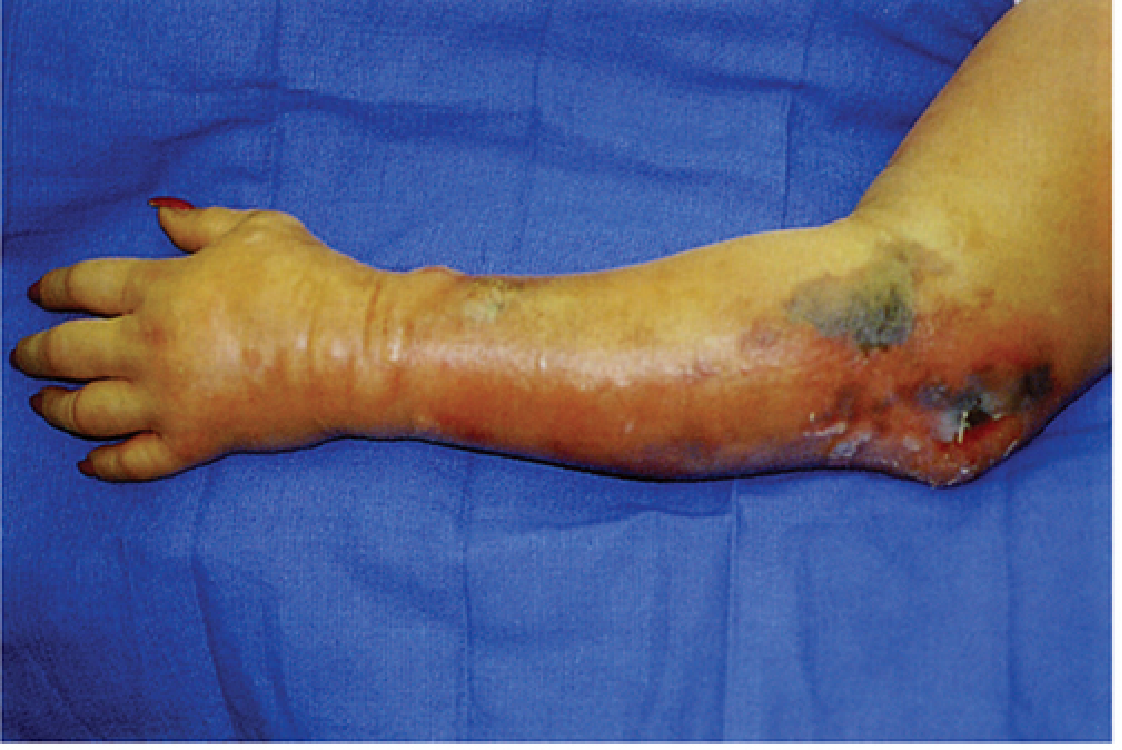
Clinical Correlation of Pathogenesis
| Feature | Pathogenic Basis |
|---|---|
| Severe pain (earliest symptom) | Edema under pressure, toxin-mediated tissue damage |
| Tense, shiny skin; pallor | Massive edema from increased vascular permeability (alpha-toxin) |
| Crepitus (gas in tissue) | CO₂ + H₂ from bacterial fermentation - a late sign |
| Thin, brown, sweet/musty-smelling exudate | Muscle/protein breakdown products |
| Hemorrhagic bullae, discoloration | Vascular damage, hemolysis |
| Paucity of pus | Leukocyte destruction by alpha and theta toxins |
| Rapid systemic deterioration | Circulating alpha-toxin → hemolysis, shock, organ failure |
Spontaneous Gas Gangrene (Special Variant)
Caused primarily by C. septicum, which is more aerotolerant. It occurs without any external wound - the organism enters the bloodstream from a GI source (often a colonic carcinoma or neutropenic enterocolitis) and seeds damaged or ischemic muscle. Bacteremia typically precedes cutaneous manifestations by several hours, so blood cultures are critical when this is suspected.
Key Pathogenesis Summary
Spore contamination of devitalized wound
↓
Low redox potential → Spore germination → Vegetative multiplication
↓
Gas production (CO₂, H₂) → Anaerobic environment maintained
↓
Alpha-toxin (lecithinase) + Theta-toxin production
↓
Cell membrane destruction → Myonecrosis + Hemolysis + PMN lysis
↓
Collagenase + Hyaluronidase → Spread along tissue planes (up to 10 cm/h)
↓
Leukocyte block → No local defense → Unimpeded spread
↓
Systemic alpha-toxin absorption → Shock → Renal failure → Death
Sources: Harrison's Principles of Internal Medicine 22e; Sherris & Ryan's Medical Microbiology 8e; Bailey & Love's Short Practice of Surgery 28e; Campbell's Operative Orthopaedics 15e (2026); Pye's Surgical Handicraft 22e
This is a shared conversation. Sign in to Orris to start your own chat.