Development of lung and associated anamolies full details for md exam answer 15 marks from fishman and Murray
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have extensive, detailed content from both Fishman's and Murray & Nadel's. Let me compile the comprehensive MD exam answer.
Development of the Lung and Associated Anomalies
[15 Marks — MD Examination Answer]
Sources: Fishman's Pulmonary Diseases and Disorders, 2-Volume Set (9781260473940) & Murray & Nadel's Textbook of Respiratory Medicine, 2-Volume (9780323655873)
I. INTRODUCTION
The human lungs serve as the primary respiratory organ with an estimated gas-exchange surface area of approximately 130 m². Gas exchange is carried out by ~480 million alveoli. Because gas exchange is required immediately at birth, developmental patterns leading to a functional lung are key to survival. Defects in lung development cause mortality and morbidity not only in the neonatal and pediatric period but often lead to respiratory deficiency in adult life, including predisposition to COPD.
Lung development is visible from approximately 4 postconceptual weeks (PCW 4) with the appearance of the trachea and paired lung buds, followed by an elaborate programme of branching morphogenesis. It is estimated that 95% of the final alveolar surface area is added postnatally, and new alveoli continue forming into the early twenties in humans.
II. STAGES OF LUNG DEVELOPMENT
Lung development is classically divided into five stages, defined by distinct morphological and cellular events:
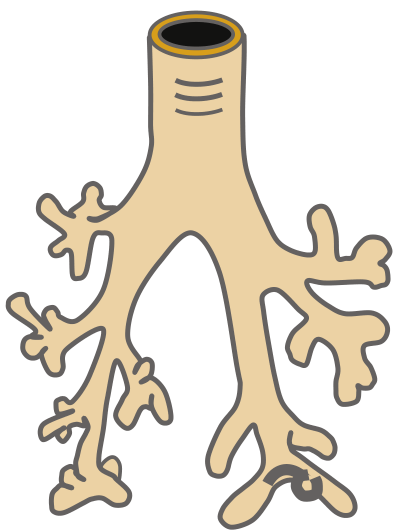
1. Embryonic Stage (PCW 4–7)
- The lung originates from the laryngotracheal groove of the endoderm at the fourth pharyngeal arch.
- A small number of endoderm-derived ventral foregut epithelial cells become specified to the respiratory fate, marked by expression of Nkx2-1 (the earliest known respiratory epithelial marker).
- The respiratory diverticulum outpouches from the foregut → forms the tracheal bud → separates from the esophagus.
- The tracheal bud branches into two primary bronchi (left and right), which divide into secondary/lobar bronchi by 5 weeks:
- Right: 3 lobar bronchi (upper, middle, lower)
- Left: 2 lobar bronchi (upper, lower)
- The splanchnic mesoderm provides muscular and cartilaginous coverings, forms visceral pleura; somatic mesoderm forms the parietal pleura.
- Left-right asymmetry of the lung buds is already specified at this stage.
2. Pseudoglandular Stage (PCW 5–16)
- The lung grows laterally and caudally on exposure to splanchnic mesoderm.
- Conducting airways progressively branch until terminal bronchioles (16th–25th generation) are formed by 16 weeks.
- Mesenchymal tissue differentiates into ciliated columnar epithelial cells, goblet cells, and eventually type II pneumocytes.
- On histology, lung resembles an exocrine gland (hence "pseudoglandular").
- This stage marks the transition between conducting and respiratory airways.
- No gas exchange is possible — a fetus born at this stage cannot survive.
3. Canalicular Stage (PCW 16–26)
- Epithelium differentiates into type I pneumocytes (alveolar cells) — the primary structural cells for gas exchange.
- Extensive angiogenesis leads to capillary network formation surrounding alveolar cells.
- Lamellar bodies form within type II pneumocytes — primary sites for surfactant storage.
- Conducting airways elongate and grow in diameter, forming a canalicular appearance.
- By the end of this stage, gas exchange becomes theoretically possible — survival is feasible with intensive support from ~24 weeks.
4. Saccular Stage (26 weeks to term)
- Progressive maturation of type I and type II pneumocytes; decrease in interstitial tissue forms air spaces called primary sacculi — the location of future gas exchange.
- Exponential growth in primary sacculi with formation of primary septa between each saccular unit.
- Primary septa contain a double-layered capillary network.
- Mesenchymal tissue further differentiates into smooth muscle and elastic fibres.
- Fetal lung fluid plays an important role: the lung epithelium is predominantly in secretory mode, generating distending pressure that stimulates growth factors and cellular proliferation.
- At birth, the epithelium switches to absorptive mode (via alterations in Na⁺/Cl⁻ transporter channels) to facilitate rapid fluid clearance and surfactant maturation.
5. Alveolar Stage (PCW 36 → young adulthood)
- The primary goal is alveologenesis — expansion of the gas-exchange surface area.
- New septa rise from alveolar walls into the lumen. Each nascent septum has flat AT1 cells and interspersed AT2 cells overlying a dense capillary network.
- Smooth muscle actin-positive myofibroblasts form a fishnet-like network; their contraction drives secondary septum formation (as described by Dr. Embry in 1970 and confirmed by 3D reconstructions).
- ~95% of final alveolar surface area is constructed postnatally.
- Although conventionally thought complete by 6–7 years, advanced imaging shows new alveoli continue forming into the early 20s.
- Early exposure to pollution, secondhand smoke, or e-cigarette aerosols truncates alveologenesis, predisposing to adult-onset COPD.
III. FETAL LUNG FLUID
- Fetal lung fluid is produced by the lung epithelium and distends the airways, providing the mechanical stimulus for lung growth.
- Oligohydramnios, decreased lung fluid, or absent fetal breathing movements → pulmonary hypoplasia.
- At birth, the epithelium switches from secretory to absorptive function; failure leads to transient tachypnea of the newborn (TTN) and respiratory distress syndrome (RDS).
IV. MOLECULAR MECHANISMS OF LUNG DEVELOPMENT
A complex interaction of growth factors and signalling molecules governs morphogenesis at each stage. Dysregulation of any pathway results in aberrant lung development:
| Molecule | Role |
|---|---|
| HNF3β, Hoxa3, Hoxa5 | Development of laryngeal-tracheal groove in foregut endoderm |
| Nkx2-1 (TTF-1) | Earliest respiratory epithelial marker; controls FGF10 |
| FGF10 (from mesenchyme) | Stimulates foregut endoderm to initiate lung branching; elevated activity → branching at tip; inhibition → arrest of branching |
| Sonic Hedgehog (Shh) | Controls FGF10; null mutations → tracheal agenesis and TEF |
| Retinoic Acid Receptor (RAR) | Co-regulates FGF10 |
| BMP family | Lung growth, vasculogenesis, alveolarization |
| EGF, PDGF, VEGF | Vasculogenesis, smooth muscle formation |
| Surfactant protein-C | Alveolarization |
| KGF/FGF7, PDGFB | Associated with CPAM when overexpressed |
Null mutations in Shh/FGF10/RAR pathways → tracheal agenesis, tracheoesophageal fistula, pulmonary hypoplasia.
V. DEVELOPMENTAL ANOMALIES OF THE LUNG
Developmental disorders are classified into airway, parenchymal, and vascular disorders (Fishman's Fig. 105-1).
A. AIRWAY DISORDERS
1. Tracheal Agenesis
- Results from arrest of the lung bud in the embryonal stage.
- Complete absence or interruption of the trachea — generally incompatible with life.
- Floyd's classification (1962):
- Type I: Absence of proximal trachea + distal tracheoesophageal fistula (TEF).
- Type II (most common, 50–60%): Complete tracheal agenesis; normal bifurcating bronchi fused at carina; esophago-carinal or esophago-bronchial fistula.
- Type III: Complete tracheal agenesis; individual primary bronchi branch from the esophagus.
- Prevalence: 1:50,000; male predominance (M:F = 2:1).
- Associated with VACTERL and TACRD syndromes.
2. Tracheoesophageal Fistula (TEF)
- Incidence: 1 in 3,500 live births; frequently with esophageal atresia.
- Part of VACTERL syndrome (Vertebral, gastrointestinal Atresia, Cardiac, Tracheoesophageal, Renal, Limb abnormalities).
- Results from failure of separation of the common foregut tube into trachea and esophagus.
- Surgically correctable by ligation; repair of esophagus may be complex depending on gap length.
3. Bridging Bronchus
- Type I: Right bridging bronchus originates from left main bronchus → supplies right middle and lower lobes; right upper lobe bronchus from carina.
- Type II: Right bridging bronchus from left main bronchus → supplies entire right lung; right main bronchus ends in a blind pouch.
4. Bronchogenic Cyst
- Derived from the lung bud of the foregut in the embryonal stage.
- Fluid-filled blind cystic pouch in mediastinum or lung parenchyma.
- Location depends on timing: Early defect → mediastinal cyst (paratracheal, subcarinal, paraesophageal, hilar); late defect → intrapulmonary cyst (lower lobes).
- Incidence: 1 in 42,000–68,000 hospital admissions; male predilection.
- Symptoms: Often asymptomatic; compression → SVC syndrome, dyspnea, dysphagia; infection → cough, purulent expectoration, fever, hemoptysis (82% of symptomatic cases).
- Imaging: CT chest (modality of choice) — density ranges from water to soft tissue; no contrast enhancement; "milk of calcium" appearance possible.
- Histopathology: Respiratory ciliated pseudostratified columnar epithelium; cartilage, smooth muscle, bronchial glands in cyst wall.
- Management: Asymptomatic — conservative; symptomatic or large — surgical resection.
B. PARENCHYMAL DISORDERS
5. Congenital Pulmonary Airway Malformation (CPAM)
(Previously: Congenital Cystic Adenomatoid Malformation / CCAM)
- Most common congenital lung mass.
- Results from abnormal branching morphogenesis.
- Connected to normal tracheobronchial tree; blood supply from pulmonary circulation.
- Incidence: 1 in 10,000–35,000 live births.
- Pathogenesis: Overexpression of KGF/FGF7 and PDGFB implicated.
Stocker Classification (5 Types):
| Type | Origin | Characteristics |
|---|---|---|
| 0 | Trachea/primary bronchus | Often lethal |
| 1 (most common, 50–70%) | Distal bronchus | Single large cyst 3–10 cm; significant mass effect |
| 2 (15–30%) | Terminal bronchioles | Evenly spaced small cysts (1–2 cm); ± solid components; associated with renal agenesis, cardiac defects, GI atresia |
| 3 | Acinar level | Microcystic/solid; poor prognosis |
| 4 | Peripheral acinar/alveolar | Large peripheral cysts; risk of malignant transformation (pleuropulmonary blastoma) |
- Management: Fetal interventions when impairing normal lung development or cardiac function; typically resected after birth due to risk of malignant transformation (though practice is evolving toward observation with serial imaging in asymptomatic patients).
6. Pulmonary Sequestration
- Non-functioning lung tissue that does not connect to the tracheobronchial tree; blood supply from systemic circulation (aberrant aortic artery).
- Rarer than CPAM.
- Types:
- Intralobar (majority): Within a normal lobe; shares visceral pleura; presents with recurrent infections.
- Extralobar: Outside normal lung; independent visceral pleural covering; associated with other anomalies.
- Hybrid: Histologic features of CPAM but systemic blood supply.
- Imaging: CT angiography — feeding systemic artery from descending aorta is the distinguishing feature.
- Management: Surgical resection in symptomatic patients; conservative management in asymptomatic adults.
7. Congenital Lobar Emphysema (CLE)
- Hyperinflation of one or more lobes (most often right upper or middle lobe).
- Causes: Bronchomalacia, extrinsic compression, mucus plugging.
- CXR: Hyperlucent, overinflated lobe with compression atelectasis of adjacent lobes + mediastinal shift.
- CT chest: Confirms hyperinflation and compression.
- Management: Symptomatic infants/children → surgical resection; asymptomatic → conservative with surveillance.
8. Pulmonary Hypoplasia
- Arrested lung development; defined as lung volume:body weight ratio below the 10th percentile for age.
- Pathological criterion: Radial alveolar count ≤ 4.1 (radial alveolar count = number of alveoli from center of respiratory bronchiole to nearest septal division transected by a perpendicular line).
- Historical definition: Lung weight:body weight < 0.12.
- Causes:
- Oligohydramnios (decreased amniotic fluid → efflux of lung fluid → decreased distending pressure → inhibited growth)
- Congenital Diaphragmatic Hernia (CDH): Abdominal organs compress lung during pseudoglandular phase → inhibits branching morphogenesis; CDH affects 1 in 3,500 live births.
- Potter syndrome (renal agenesis → oligohydramnios); lungs and kidneys share branching morphogenesis signalling.
- Decreased fetal breathing movements (neuromuscular disease).
- Premature rupture of membranes (PROM) during canalicular phase.
C. VASCULAR DISORDERS
9. Pulmonary Artery Sling (PAS)
- Anomalous origin of the left pulmonary artery from the right pulmonary artery → it passes between the trachea and esophagus, compressing the posterior trachea.
- Associated with complete tracheal rings in up to 80% of cases.
- Type I: Located immediately above the carina; compresses right main bronchus; associated with tracheal bronchus; tracheobronchomalacia → hyperinflation of right lung.
- Type II (more inferior, at T5–T6): Associated with abnormally low horizontal carina and bridging bronchus; subdivided into:
- IIA: Tracheal bronchus at usual carina supplies RUL; bridging bronchus from left main bronchus supplies RML and RLL.
- IIB: No right upper lobe bronchus; entire right lung supplied by bridging bronchus. Both type II variants strongly associated with long-segment tracheal stenosis.
- Additional associations: Right-sided pulmonary hypoplasia, scimitar syndrome, foregut abnormalities.
- Imaging: CXR (type I — prominent vasculature; type II — T-shaped carina, distal trachea visibility poor, RLL atelectasis); CT/MRI angiography; 3D reformation for surgical planning.
- Management: Asymptomatic type I → monitor; symptomatic → reimplant left PA anterior to trachea; type II — slide tracheoplasty for long-segment tracheal stenosis.
10. Scimitar Syndrome (Congenital Venolobar Syndrome)
- Anomalous pulmonary venous drainage of right lung into the inferior vena cava → creates the characteristic "scimitar" sign on CXR.
- Associated with right lung hypoplasia, dextrocardia, and systemic arterial supply.
VI. CONGENITAL LUNG DISEASES FROM MURRAY & NADEL'S — ADDITIONAL PERSPECTIVE
| Condition | Timing | Key Mechanism |
|---|---|---|
| Tracheoesophageal fistula | Embryonic | Failure of foregut separation |
| Tracheal atresia | Embryonic | Arrest of lung bud |
| Bronchogenic cyst | Embryonic | Abnormal budding of ventral foregut |
| CPAM | Pseudoglandular | Abnormal branching morphogenesis |
| Pulmonary sequestration | Pseudoglandular | No connection to tracheobronchial tree; systemic blood supply |
| Lung hypoplasia | Canalicular/Pseudoglandular | CDH compression, oligohydramnios |
| Surfactant deficiency/RDS | Canalicular/Saccular | Failure of type II pneumocyte maturation |
| BPD | Postnatal | Preterm birth → arrested alveologenesis |
| Childhood ILD (chILD) | Alveolar | Mutations in surfactant proteins |
VII. BRONCHOPULMONARY DYSPLASIA (BPD)
- A common consequence of preterm birth (alveologenesis not complete).
- Results in poor gas exchange in infancy; frequently persists into adulthood.
- Associated with increased susceptibility to respiratory infections.
- Reduced lung capacity due to abnormal early lung growth → premature decline of respiratory function, predisposing to adult-onset COPD.
VIII. SURFACTANT DEFICIENCY
- Surfactant (stored in lamellar bodies of type II pneumocytes from the canalicular stage) is critical to reduce alveolar surface tension.
- Mutations in surfactant proteins (SP-B, SP-C, ABCA3 gene) → childhood interstitial lung diseases (chILD).
- Preterm neonates lack adequate surfactant → Respiratory Distress Syndrome (RDS/Hyaline Membrane Disease).
Summary Table: Five Stages of Lung Development
| Stage | Timing | Key Events | Clinical Significance |
|---|---|---|---|
| Embryonic | PCW 4–7 | Lung bud, tracheal-bronchial tree to lobar bronchi | TEF, tracheal agenesis, bronchogenic cysts |
| Pseudoglandular | PCW 5–16 | Conducting airways to terminal bronchioles; no gas exchange | CPAM, sequestration; CDH compresses lungs here |
| Canalicular | PCW 16–26 | Type I/II pneumocytes; capillary network; lamellar bodies/surfactant | Limit of viability ~24 wks; RDS; oligohydramnios → hypoplasia |
| Saccular | 26 wks–term | Primary sacculi; primary septa; double-layer capillaries | Preterm birth → RDS, TTN |
| Alveolar | 36 wks–early 20s | Secondary septation; 95% of surface area postnatal | BPD (preterm arrest); COPD predisposition from early toxic exposure |
References:
- Fishman's Pulmonary Diseases and Disorders, 2-Volume Set — Chapter 105: Developmental Disorders of the Lung (pp. 1856–1870)
- Murray & Nadel's Textbook of Respiratory Medicine, 2-Volume — Chapter 2: Stages of Lung Development with Cellular and Molecular Mechanisms (pp. 63–70)
This is a shared conversation. Sign in to Orris to start your own chat.