Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive information from Murray & Nadel's Textbook of Respiratory Medicine and other authoritative sources to provide a detailed answer. Let me compile the full explanation.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Definition
ARDS is a syndrome of acute hypoxemic respiratory failure defined by the Berlin criteria (2012):
- Onset within 1 week of a known clinical insult or new/worsening respiratory symptoms
- Bilateral opacities on chest imaging not fully explained by effusions, lobar/lung collapse, or nodules
- Respiratory failure not fully explained by cardiac failure or fluid overload
- Hypoxemia stratified by PaO2/FiO2 ratio on PEEP ≥5 cmH2O:
- Mild: PaO2/FiO2 201-300
- Moderate: PaO2/FiO2 101-200
- Severe: PaO2/FiO2 ≤100
Inciting Causes
ARDS arises from two broad categories of lung insult:
| Direct (Pulmonary) Injury | Indirect (Nonpulmonary) Injury |
|---|---|
| Pneumonia (bacterial, viral - e.g. influenza, COVID-19) | Sepsis (most common overall) |
| Aspiration of gastric contents | Trauma, polytrauma |
| Pulmonary contusion | Multiple blood transfusions (TRALI) |
| Smoke/toxic inhalation | Pancreatitis |
| Near-drowning | Burns |
| Fat embolism | Drug overdose |
Sepsis, trauma, aspiration, and massive transfusion carry the highest individual risk. Multiple simultaneous risk factors greatly amplify likelihood of ARDS. Chronic alcoholism doubles the relative risk.
- Murray & Nadel's Textbook of Respiratory Medicine, Table 134.2
Core Pathophysiological Mechanism
The central event in ARDS is loss of integrity of the alveolar-capillary barrier, leading to noncardiogenic, protein-rich (exudative) pulmonary edema - unlike cardiogenic edema which is driven by elevated left-sided pressures.
1. Alveolar-Capillary Barrier Disruption
Both the microvascular endothelium and the alveolar epithelium are damaged:
-
Endothelial injury: Loss of pulmonary vascular endothelial barrier integrity is both necessary and sufficient for ARDS to develop. When this barrier is breached, protein-rich plasma fluid floods the interstitium.
-
Epithelial injury: Type I pneumocytes (covering ~95% of alveolar surface) are particularly vulnerable. Their destruction disrupts barrier integrity AND eliminates the Na+/K+ ATPase-driven fluid reabsorption mechanism that normally keeps alveoli dry. Type II pneumocyte injury further impairs surfactant production.
-
Mechanisms of cell death include necrosis, apoptosis, coagulation-mediated injury, and mechanical stretch.
-
Murray & Nadel's Textbook of Respiratory Medicine, p. 3146
2. Neutrophil-Mediated Injury (Central Role)
One of the earliest signs of ARDS - even before overt hypoxemia - is transient leukopenia from sequestration of neutrophils in the pulmonary microvasculature.
The sequence:
- Neutrophil sequestration: Pulmonary capillaries are narrower than the diameter of an average neutrophil. Activated neutrophils become "stiff" through actin cytoskeletal changes and cannot deform to pass through, trapping them in capillaries.
- Transendothelial migration: Sequestered neutrophils migrate into the interstitium and alveolar space, where they release cytotoxic compounds:
- Reactive oxygen species (ROS) - oxidative damage to membranes and proteins
- Proteolytic enzymes (leukocyte elastase, matrix metalloproteinases) - degrade structural proteins and surfactant apoprotein-A
- Cationic peptides (defensins) - direct cytotoxicity
- Eicosanoids, cytokines (TNF-α, IL-1β) - amplify the inflammatory cascade
- Neutrophil extracellular traps (NETs) - DNA-histone nets that trap pathogens but also damage host tissue
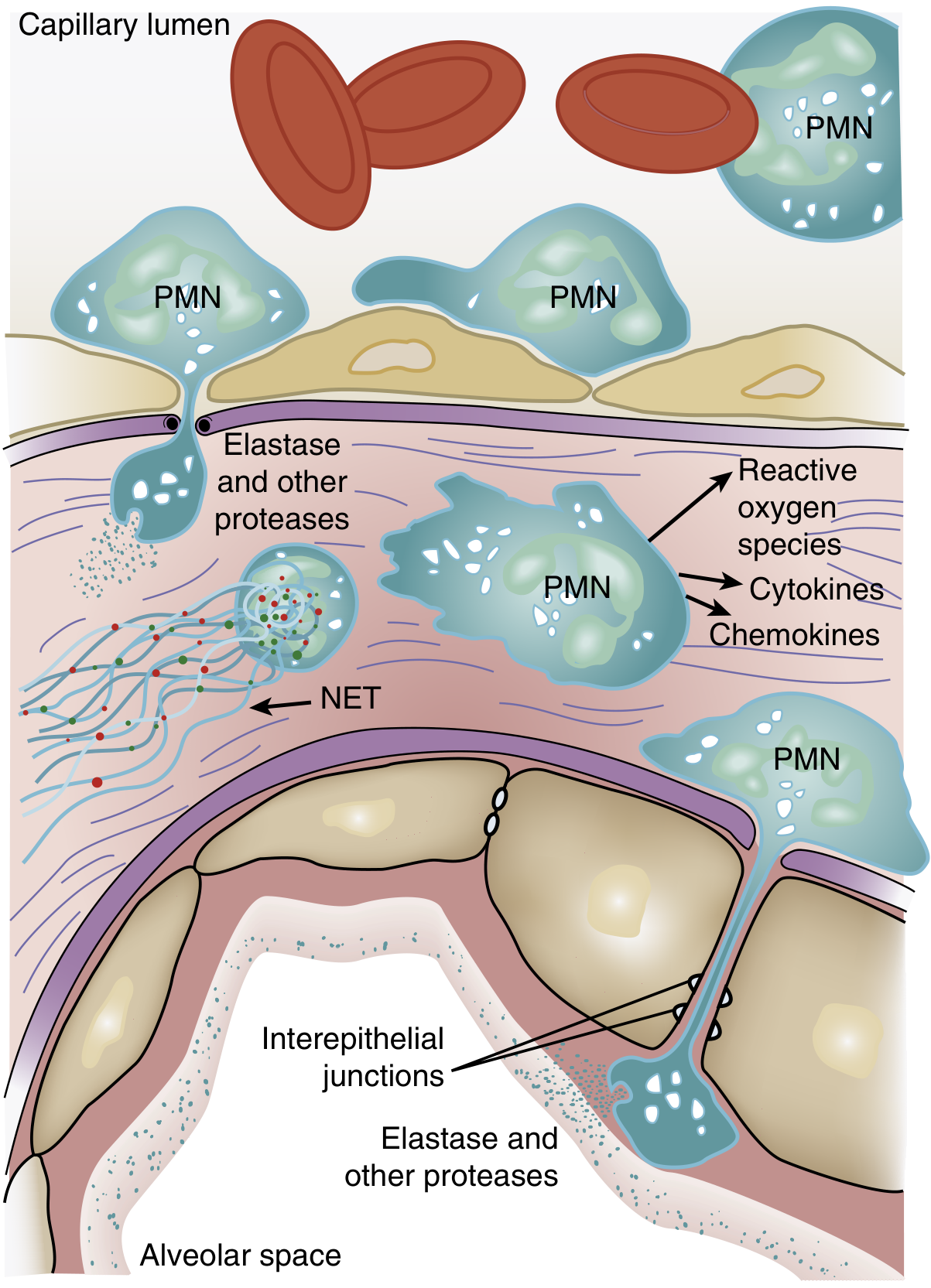
Figure: Role of neutrophils in ARDS pathogenesis - Murray & Nadel's Textbook of Respiratory Medicine
- Murray & Nadel's Textbook of Respiratory Medicine, pp. 3147-3148
3. Cytokine Storm and Inflammatory Mediators
A self-amplifying inflammatory cascade is set off:
- TNF-α and IL-1β: Released early by alveolar macrophages after sensing DAMP/PAMP signals. These cytokines activate neutrophils and upregulate adhesion molecules on endothelium, promoting further neutrophil recruitment.
- IL-8 (CXCL8): A potent neutrophil chemoattractant; elevated in BAL fluid of ARDS patients.
- Platelet-activating factor (PAF): Increases vascular permeability.
- Reactive oxygen species: Generated by activated neutrophils and macrophages, causing widespread membrane lipid peroxidation.
- Phospholipase A2 (elevated in pancreatitis-associated ARDS): Degrades surfactant and increases vascular permeability.
- In pancreatitis: Proinflammatory cytokines including TNF-α and IL-8 augment neutrophil recruitment to the lungs.
4. Surfactant Dysfunction
- Activated neutrophil elastase degrades surfactant protein-A.
- The ratio of large (active) to small (inactive) surfactant aggregates falls, due to decreased production and increased conversion.
- Plasma proteins leaking through the damaged barrier (fibrin, fibrinogen, albumin) directly inhibit surfactant function.
- Result: increased alveolar surface tension → alveolar collapse (atelectasis) and further V/Q mismatch.
5. Coagulation Abnormalities
- Platelet and coagulation pathway activation leads to fibrin-platelet microthrombi in the pulmonary microvasculature.
- This worsens perfusion, contributes to pulmonary hypertension, and increases dead space.
- Procoagulant + antifibrinolytic shifts in the alveolar space promote intraalveolar fibrin deposition, compounding surfactant dysfunction.
- Elevated pulmonary dead space fraction (often >2x normal minute ventilation needed) reflects poorly perfused lung units.
6. Angiopoietin Pathway
- Angiopoietin-2 (Ang2): Released from endothelial cells, acts as a competitive antagonist of Ang1 at the Tie2 receptor. Elevated Ang2 promotes vascular leak and destabilizes the endothelial barrier. Genetic variants in Ang2 are associated with higher plasma levels and increased ARDS risk.
- Angiopoietin-1 (Ang1): Counter-regulatory; protects barrier integrity. Exogenous Ang1 reverses Ang2-induced vascular leak in vitro.
Pathological Phases (Diffuse Alveolar Damage - DAD)
ARDS classically progresses through three overlapping stages:
| Phase | Timing | Histology |
|---|---|---|
| Exudative | Days 1-7 | Alveolar/interstitial edema, hemorrhage, hyaline membrane formation (precipitated plasma proteins + fibrin + necrotic debris), neutrophil infiltration, capillary congestion, atelectasis |
| Proliferative | Days 7-21 | Reorganization of hyaline membranes, type II pneumocyte hyperplasia (attempting repair), early fibrosis, decreased neutrophils, reduced pulmonary edema |
| Fibrotic | >2-3 weeks | Pulmonary fibrosis in a subset; obliteration of pulmonary capillaries; interstitial and alveolar collagen deposition. Collagen synthesis (N-terminal procollagen-III) detectable in BAL fluid within 24h of onset |
Note: DAD is confirmed on autopsy in only ~45-50% of ARDS cases. Non-DAD ARDS may reflect infectious pneumonia, diffuse alveolar hemorrhage, acute eosinophilic pneumonia, or drug pneumonitis.
- Murray & Nadel's Textbook of Respiratory Medicine, pp. 3146, 3075-3076
Physiological Consequences
| Consequence | Mechanism |
|---|---|
| Severe hypoxemia | Right-to-left intrapulmonary shunting through fluid-filled alveoli; low V/Q units |
| Increased dead space | Microthrombi and vascular compression → ventilated but unperfused units; minute ventilation rises to 12-16 L/min |
| Decreased lung compliance | Loss of ventilated lung volume; surfactant failure; alveolar edema |
| Pulmonary hypertension | Hypoxic vasoconstriction + intravascular fibrin + mechanical compression by positive-pressure ventilation |
| Impaired alveolar fluid clearance | Hypoxia suppresses apical Na+ channels and basolateral Na+/K+ ATPase on type II pneumocytes, blocking active fluid removal |
Summary Flowchart
Triggering insult (sepsis / aspiration / trauma / pneumonia)
↓
Alveolar macrophage activation → TNF-α, IL-1β, IL-8
↓
Neutrophil sequestration in pulmonary capillaries
↓
PMN transmigration → ROS, elastase, NETs, cytokines
↓
Alveolar-capillary barrier disruption
(endothelial + epithelial injury)
↓
Protein-rich exudate floods alveoli + interstitium
↓
Surfactant dysfunction → alveolar collapse
Coagulation activation → microthrombi
Na+/H2O clearance failure
↓
Hypoxemia, ↓ compliance, ↑ dead space, pulmonary HTN
↓
ARDS (Diffuse Alveolar Damage)
Sources:
- Murray & Nadel's Textbook of Respiratory Medicine (2-Volume), Chapters 133-134
- Goldman-Cecil Medicine International Edition, Chapter 91
- Sabiston Textbook of Surgery, Chapter on Critical Care
This is a shared conversation. Sign in to Orris to start your own chat.