Myocardial infraction
Reading File
Finding Sources
Searching PubMed
"myocardial infarction"[MeSH Terms] AND management
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive information from authoritative textbooks. Let me compile the full answer.
Myocardial Infarction (Heart Attack)
Definition
Myocardial infarction (MI) is the death of cardiac muscle due to prolonged ischemia. In the United States, roughly 800,000 individuals experience an MI each year (nearly one MI every 40 seconds), causing almost 400,000 deaths annually. The major underlying cause is atherosclerosis.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 511
Pathogenesis
Coronary Arterial Occlusion
The following sequence underlies most MIs:
- An atheromatous plaque is eroded or suddenly disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces, exposing subendothelial collagen and necrotic plaque contents to blood.
- Platelets adhere, aggregate, and release thromboxane A2, ADP, and serotonin - causing further platelet aggregation and vasospasm.
- Activation of coagulation by tissue factor adds to the growing thrombus.
- Within minutes, the thrombus can fully occlude the coronary artery lumen.
When angiography is performed within 4 hours of MI onset, coronary thrombosis is demonstrated in almost 90% of cases. This has therapeutic implications: early thrombolysis and/or angioplasty can be highly successful in limiting myocardial necrosis.
In approximately 10% of cases, MI occurs without typical coronary atherothrombosis. Other mechanisms include:
-
Vasospasm (e.g., cocaine or ephedrine use)
-
Embolism (from left atrial mural thrombus in atrial fibrillation, infective endocarditis vegetations, prosthetic material)
-
Disorders of small intramural coronary vessels, sickle cell disease, vasculitis, aortic stenosis
-
Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 511-512
Progression of Necrosis
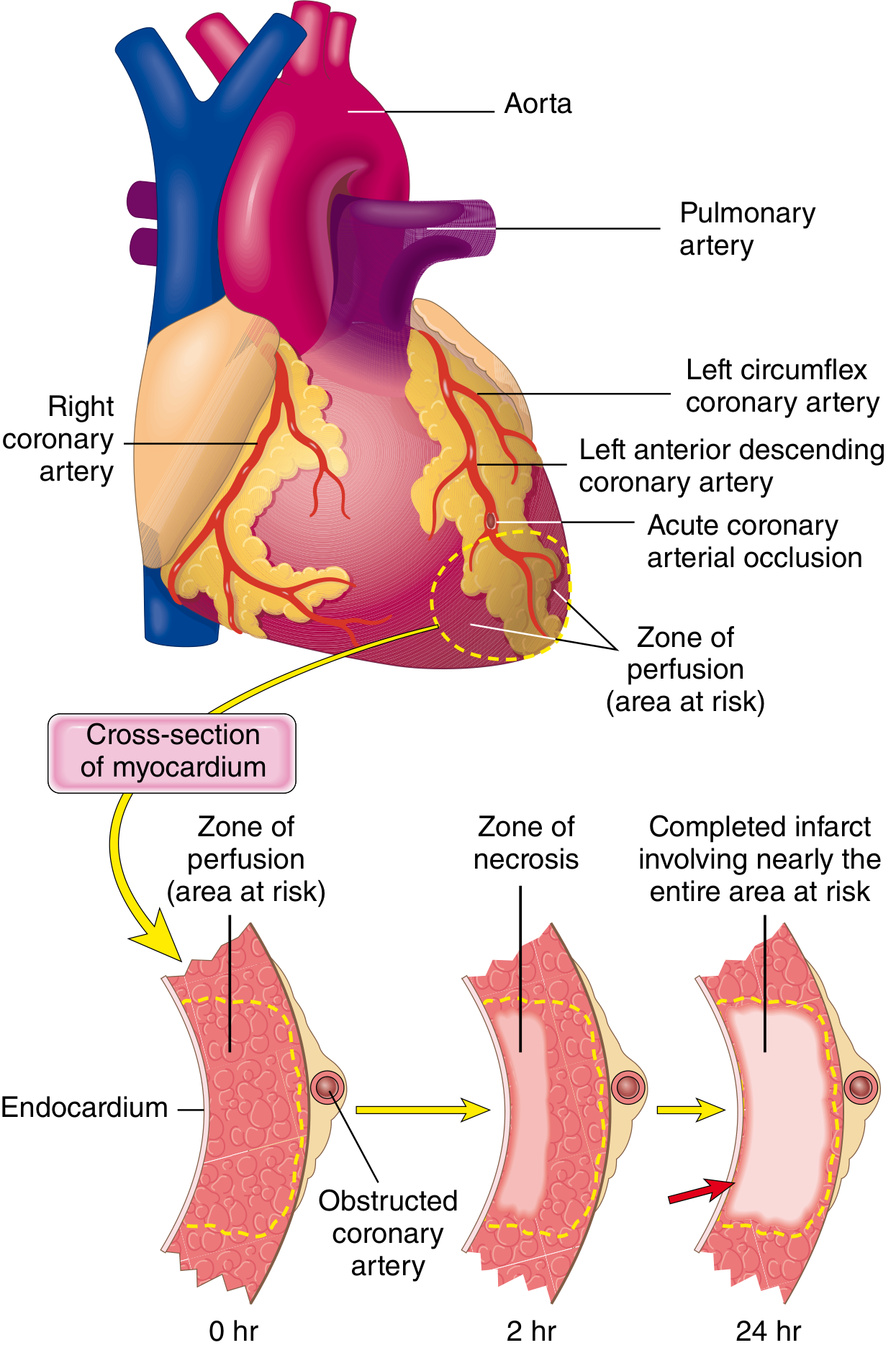
Fig. 12.10 - Progression of myocardial necrosis after coronary artery occlusion. Necrosis begins in the subendocardial zone and progresses outward as a wavefront over 6-24 hours.
The timeline of ischemic events:
| Feature | Time |
|---|---|
| Onset of ATP depletion | Seconds |
| Loss of contractility | <2 minutes |
| ATP reduced to 50% of normal | 10 minutes |
| ATP reduced to 10% of normal | 40 minutes |
| Irreversible cell injury | 20-40 minutes |
| Microvascular injury | >1 hour |
Irreversible injury first occurs in the subendocardial zone - the last area to receive blood from epicardial vessels and the area exposed to the highest intramural pressures. With prolonged ischemia, a wavefront of cell death moves centripetally. An infarct usually achieves its full extent within 6 to 12 hours.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 512
Morphology and Histology
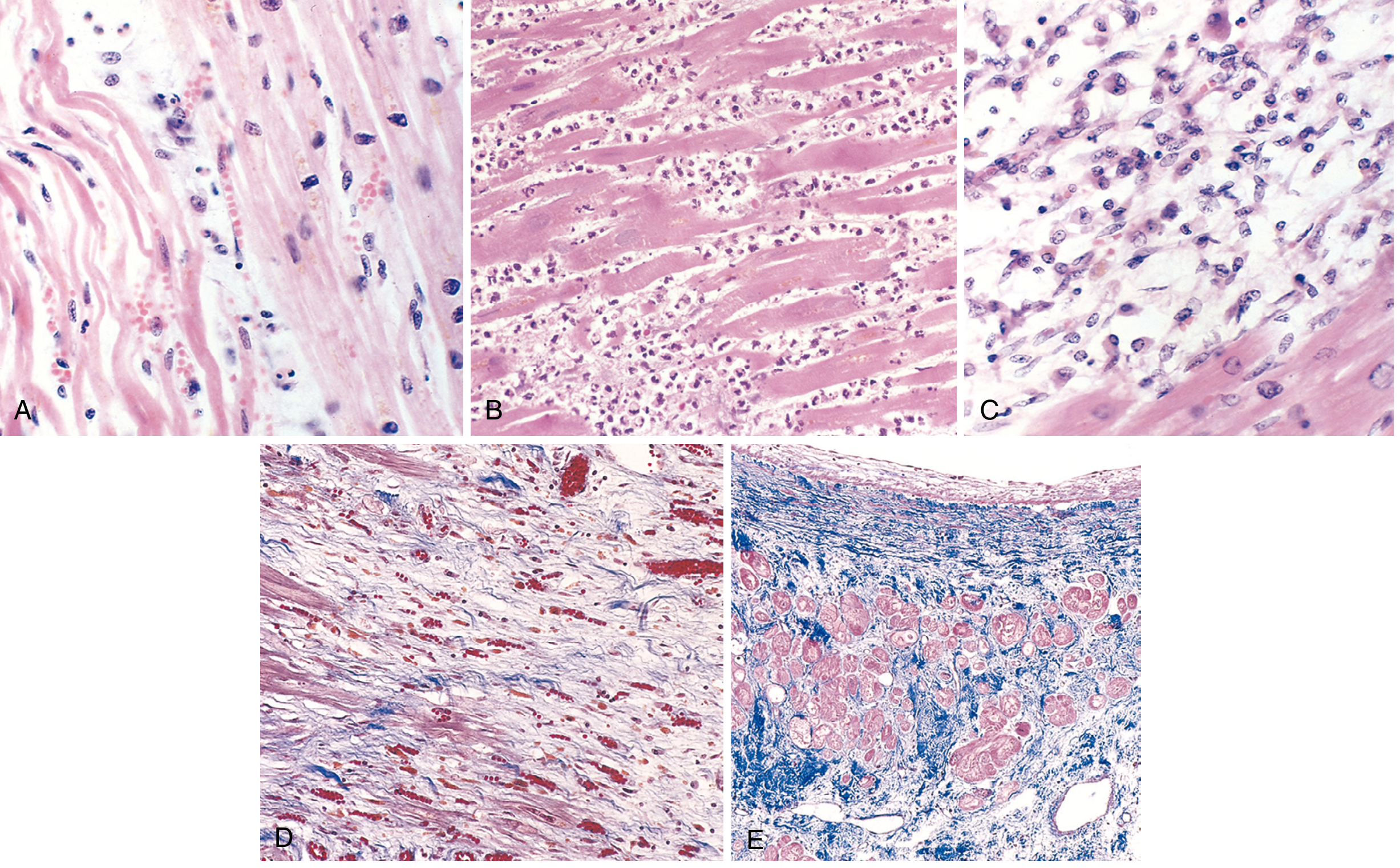
Fig. 12.13 - Microscopic features of MI and repair. (A) 1-day-old infarct: coagulative necrosis and wavy fibers. (B) 3-4 days: dense neutrophil infiltrate. (C) 7-10 days: macrophage phagocytosis of necrotic myocytes. (D) Granulation tissue with loose collagen and abundant capillaries. (E) Healed infarct: dense collagenous scar (blue, Masson trichrome).
The gross and microscopic changes evolve predictably:
| Time | Gross Changes | Microscopic Changes |
|---|---|---|
| 0-4 hours | None visible | None visible (EM: myofibril relaxation, mitochondrial swelling) |
| 4-12 hours | Dark mottling (early) | Coagulative necrosis begins; wavy myofibers |
| 12-24 hours | Dark mottling | Pyknosis, contraction bands, neutrophil infiltrate begins |
| 1-3 days | Mottling with yellow-tan infarct center | Extensive coagulative necrosis; heavy neutrophil infiltrate |
| 3-7 days | Yellow-tan center, hyperemic border | Neutrophil disintegration; macrophage phagocytosis begins |
| 7-10 days | Maximally yellow-tan, soft, depressed borders | Phagocytosis of necrotic myocytes; granulation tissue |
| 2-8 weeks | Gray-white fibrosis | Fibrous scar formation |
| >2 months | Dense fibrous scar | Complete scarring; residual myocytes show compensatory hypertrophy |
Classification
By ECG pattern:
- STEMI (ST-Elevation MI) - transmural infarction (full wall thickness); ST segment elevation in leads overlying the infarct
- NSTEMI (Non-ST-Elevation MI) - subendocardial infarction; no ST elevation, but troponin positive
By territory:
- Anterior MI - usually left anterior descending (LAD) artery occlusion
- Inferior MI - usually right coronary artery (RCA) occlusion
- Lateral MI - usually left circumflex artery occlusion
ECG Changes (Ganong's Physiology)
Three major abnormalities cause ECG changes in acute MI:
| Defect in Infarcted Cells | Current Flow | ECG Change in Leads over Infarct |
|---|---|---|
| Rapid repolarization | Out of infarct | ST segment elevation |
| Decreased resting membrane potential | Into infarct | TQ segment depression (manifested as ST elevation) |
| Delayed depolarization | Out of infarct | ST segment elevation |
The hallmark of acute MI is ST segment elevation in leads overlying the infarct. Leads on the opposite side show ST segment depression (reciprocal changes).
After days to weeks, the ST segment abnormalities subside. Dead muscle becomes electrically silent. The hallmark of old MI is the appearance or deepening of Q waves - abnormal Q waves are 0.04 s or wider and/or deeper than 25% of the R wave in the same lead.
- Ganong's Review of Medical Physiology, p. 534
Biomarkers
- Cardiac Troponins T and I (cTnT, cTnI): Most clinically useful. Rise in 2-4 hours, peak at 24-48 hours, remain elevated for 7-10 days. With reperfusion, may peak earlier ("washout" pattern).
- CK-MB: Rises in 4-6 hours, peaks at 24 hours, normalizes in 48-72 hours - useful for detecting reinfarction.
- Myoglobin: Earliest to rise (1-4 hours) but non-specific.
Importantly, low-level troponin elevation can occur in other conditions (CHF, pulmonary embolism, renal failure, sepsis), so serial measurements and clinical context are essential.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 517
Treatment
Immediate (STEMI) - "Time is Myocardium"
Reperfusion is the cornerstone of STEMI treatment:
- Primary PCI (percutaneous coronary intervention): Goal door-to-balloon time <90 minutes (or <120 minutes from first medical contact). Preferred when available.
- Fibrinolysis (e.g., tPA, streptokinase): If PCI not available within 120 minutes of first medical contact. Goal door-to-needle time <30 minutes.
Pharmacological therapy:
- Morphine 2-4 mg IV every 5 min - for pain relief (watch for hypotension, bradycardia)
- Oxygen - if SaO2 <90% or respiratory distress
- Nitrates - for vasodilation and relief of vasospasm
- Aspirin (162-325 mg chewed immediately) + P2Y12 inhibitor (clopidogrel, ticagrelor, or prasugrel) - dual antiplatelet therapy (DAPT)
- Anticoagulation - unfractionated heparin, LMWH, bivalirudin, or fondaparinux
- Beta-blockers (e.g., metoprolol 5 mg IV every 2-5 min x3 doses, then oral 50 mg every 6h) - reduce O2 demand, decrease risk of VF and reinfarction. Avoid in acute HF, low output state, cardiogenic shock, bradycardia, or high-degree AV block.
- ACE inhibitors / ARBs - started within 24 hours, especially with anterior MI, reduced EF, hypertension, or diabetes
- Statins - high-intensity statin therapy immediately
- Harrison's Principles of Internal Medicine 22E, p. 2162
Consequences and Complications
| Complication | Time of Onset | Notes |
|---|---|---|
| Arrhythmias (VF, VT, heart block) | Hours to days | Most common cause of death; VF most common in first hour |
| Cardiogenic shock | Hours to days | LV pump failure; mortality ~50-80% without support |
| Left ventricular failure / Pulmonary edema | Hours to days | Related to infarct size |
| Myocardial rupture (free wall) | 3-7 days | Catastrophic; pericardial tamponade |
| Ventricular septal defect (VSD) | 3-7 days | Harsh holosystolic murmur; may need surgical repair |
| Papillary muscle rupture / Mitral regurgitation | 3-5 days | Acute MR; sudden pulmonary edema |
| Pericarditis (Dressler syndrome) | Days to weeks | Autoimmune; treated with NSAIDs |
| Left ventricular aneurysm | Weeks to months | Risk of mural thrombus and systemic emboli |
| Mural thrombus + systemic embolism | Days to weeks | Risk of stroke; anticoagulate |
| Ventricular remodeling / Chronic HF | Weeks to months | Infarct expansion, hypertrophy of remaining myocardium |
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 501-502
Reperfusion Injury
Although restoring blood flow is beneficial, it can cause additional damage:
- Mitochondrial dysfunction - ischemia alters mitochondrial membrane permeability, leading to swelling and release of pro-apoptotic contents
- Myocyte hypercontracture - calcium overload during ischemia causes contraction band necrosis on reperfusion
- Oxygen free radicals - generated on reperfusion
- Reperfusion arrhythmias
Up to 50% or more of the ultimate infarct size may be attributed to reperfusion injury. Clinical trials to prevent it have not yet been successful.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 514
Recent Evidence Update
A 2025 meta-analysis (Chi et al., Eur J Prev Cardiol, PMID 39298680) found that beta-blockers for secondary prevention after MI in patients without reduced ejection fraction or heart failure may have a more limited benefit than previously assumed - an important update to the traditional blanket recommendation for post-MI beta-blockade in all patients.
A 2024 pooled cohort meta-analysis (Peek et al., Eur Heart J, PMID 39378245) highlighted that sudden cardiac death risk post-MI remains a major concern and can be stratified using individual patient data.
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease | Braunwald's Heart Disease | Harrison's Principles of Internal Medicine 22E | Ganong's Review of Medical Physiology | PubMed 2024-2025
This is a shared conversation. Sign in to Orris to start your own chat.