Thyroid in neonates
Reading File
Finding Sources
Finding Sources
Finding Sources
Searching PubMed
congenital hypothyroidism neonatal screening
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive information. Let me compile the full answer:
Thyroid in Neonates
1. Fetal Thyroid Development
The thyroid gland originates at week 3 of gestation from endodermal cells at the base of the tongue between the first and second pharyngeal pouch. The thyroid diverticulum descends to its pretracheal position between 5–7 weeks, with bilobar division at ~5 weeks. Parafollicular C-cells arise from the ultimobranchial bodies (4th and 5th pouches) and form Zückerkandl's tubercle. The thyroglossal duct degenerates by week 10 — incomplete degeneration causes thyroglossal duct cysts or pyramidal lobe.
Key developmental milestones:
| Gestational Age | Event |
|---|---|
| Week 3 | Thyroid primordium forms |
| Weeks 5–7 | Descent to pretracheal position |
| Weeks 10–12 | Hypothalamic/pituitary-vascular maturation; TRH detectable |
| Weeks 12–14 | Active iodide trapping; TH synthesis begins (TPO, NIS, Tg expressed) |
| Weeks 18–20 | TH secretion, TSH, and T4 rise significantly |
| Week 20 | T4 ~2 μg/dL |
| Term | T4 reaches adult levels (~10 μg/dL) |
| Before 16 weeks | Fetus entirely dependent on maternal TH |
Fetal T3 is negligible until week 30 due to high D3:D1 deiodinase ratio (T4 preferentially converted to reverse T3). Near term, T3 rises to ~50 ng/dL. At birth, 30–50% of T4 in cord blood is of maternal origin, providing partial protection to fetuses with congenital hypothyroidism.
2. Normal Neonatal Thyroid Physiology
Immediately after birth:
- TSH surges (neonatal TSH surge) within minutes of delivery, driven by cold exposure and TRH
- This triggers a rise in T4 conversion to T3
- T4 levels transiently rise (transient hyperthyroxinemia may reflect thermogenesis adaptation)
- TSH normalizes to adult levels within a few days via T3/T4 negative feedback
- T4 and T3 return to normal adult values within 4–6 weeks
- The hypothalamic-pituitary-thyroid (HPT) axis fully matures at 1–2 months after birth
In premature neonates (<28 weeks):
- HPT axis is immature; the physiological TSH surge is dramatically lower or absent
- FT4 and FT3 are low with paradoxically normal TSH
- Takes 3–8 weeks to reach levels similar to term infants
- Transient hypothalamic hypothyroidism of prematurity occurs in up to 50% of infants born <28 weeks
- Dopamine (used in NICU) can suppress TSH release further
3. Congenital Hypothyroidism (CH)
Incidence
- Estimated 1 in 2,000–4,000 newborns (higher with sensitive TSH cut-offs)
- Higher incidence in Asians, Hispanics, premature infants, and older mothers
- Prior to newborn screening, estimated at 1 in 7,000
Classification
Permanent CH (75–86%) — requires lifelong treatment:
- Thyroid dysgenesis (85%): agenesis, ectopy, or hypoplasia
- Dyshormonogenesis (15%): structurally normal gland with hormone synthesis defect
Transient CH — resolves within weeks to months:
- Endemic iodine deficiency (most common worldwide)
- Maternal antithyroid drug exposure
- Transfer of maternal TSH-receptor blocking antibodies
- Maternal iodine excess (e.g., amiodarone, contrast media)
- Liver hemangiomas (excess deiodinase 3 activity)
- Certain genetic defects
Central (Secondary/Tertiary) CH: 1 in 25,000–50,000 newborns — hypothalamic/pituitary deficiency
Causes by Category (Harrison's, 2025)
- Thyroid dysgenesis: 65%
- Dyshormonogenesis (inborn errors of TH synthesis): 30%
- TSH-receptor antibody mediated: 5%
- Developmental abnormalities twice as common in girls
4. Genetics of Congenital Hypothyroidism
| Defective Gene | Type | Inheritance | Consequence |
|---|---|---|---|
| TTF-1 (TITF-1) | Dysgenesis | Heterozygous LOF | Thyroid hyperplasia, choreoathetosis, pulmonary problems |
| TTF-2 (FOXE-1) | Dysgenesis | Homozygous recessive | Thyroid agenesis, choanal atresia, spiky hair |
| PAX-8 | Dysgenesis | Heterozygous LOF | Thyroid dysgenesis, kidney abnormalities |
| NKX2-1 | Dysgenesis | Heterozygous LOF | Thyroid + brain + lung abnormalities |
| NKX2-5 | Dysgenesis | Heterozygous LOF | Thyroid + heart abnormalities |
| GLIS3 | Dysgenesis | Homozygous recessive | Thyroid dysgenesis + neonatal diabetes + facial abnormalities |
| TSH receptor | Dyshormonogenesis | Homozygous recessive | Resistance to TSH |
| NIS (SLC5A5) | Dyshormonogenesis | Homozygous recessive | Inability to transport iodide |
| DUOX2/DUOXA2 | Dyshormonogenesis | AR / Heterozygous LOF | Organification defect |
| TPO | Dyshormonogenesis | Homozygous recessive | Organification defect |
| PROP-1, PIT-1 | Central | Homozygous recessive | Combined pituitary hormone deficiencies |
| IGSF1 | Central | X-linked LOF | Loss of TSH-R expression, testicular enlargement |
Dyshormonogenesis: autosomal recessive. Thyroid dysgenesis: only ~2% of cases are inherited; most are sporadic (polygenic/epigenetic).
5. Clinical Features
Early signs (often absent at birth due to maternal TH protection):
- Lethargy, increased sleep
- Prolonged neonatal jaundice
- Myxedematous facies
- Large anterior fontanelle
- Macroglossia
- Distended abdomen, umbilical hernia
- Hypothermia, hypotonia
Late signs (if untreated):
- Poor sucking and feeding difficulties
- Constipation
- Developmental delay
- Cognitive and growth retardation
- Myxedema
- Decreased activity
Important: 10% of CH infants have other congenital defects; of these, 50% have congenital heart defects.
6. Neonatal Screening
Newborn screening is universal in the US and most industrialized countries. Worldwide, ~25% of all newborns are screened.
Timing: Ideally days 2–4 of life for term infants; within 7 days for preterm infants. Children discharged on day 1 may be sampled at that time.
Screening strategies:
- TSH first (most common) → reflex T4 if TSH elevated: misses central hypothyroidism
- T4 first → reflex TSH if T4 low
- Combined TSH + FT4 ± Tg: differentiates primary vs. central causes
Filter paper heel prick → mailed to central lab. Each program has its own cut-off values.
Pitfalls:
- False-positive TSH elevations in the first 2 days of life (physiologic TSH surge)
- Transplacental maternal TSH heterophile antibodies → false-positive TSH
- Premature/acutely ill infants: late TSH rise → may be missed on first sample → many programs do a second screen
- Dopamine suppresses TSH in NICU infants
- Seasonal variation: false-positive rate higher in winter (0.9%) vs. summer (0.6%)
Confirmatory testing (serum): TSH + free T4 (or total T4), age-specific reference intervals. Further workup includes radionuclide thyroid scan, thyroid ultrasound, and serum thyroglobulin — but must not delay treatment.
7. Treatment
- Levothyroxine (LT4) is the treatment of choice
- Goal: normalize serum TSH and raise T4 to age-appropriate levels
- If started within the first 2 weeks of life → prevents mental retardation in >90% of children
- Dose is weight-based and adjusted as the infant grows
8. Neonatal Graves Disease
If the mother has Graves disease, stimulatory TSH-receptor antibodies (TSI/TRAb) can cross the placenta and stimulate the fetal/neonatal thyroid, causing:
- Fetal/neonatal hyperthyroidism
- Thyrotoxicosis, tachycardia, goiter, advanced bone age
Measurement of TSH-receptor antibodies (thyroid-stimulating Ig assay) during pregnancy is useful for risk assessment. The condition is transient as maternal antibodies are cleared.
Sources:
- Creasy & Resnik's Maternal-Fetal Medicine, Ch. 61
- Tietz Textbook of Laboratory Medicine, 7th Ed., Ch. 26
- Harrison's Principles of Internal Medicine, 22nd Ed. (2025), Ch. 395
Meromelia in neonates causes
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Searching Images
meromelia phocomelia limb reduction defect neonate
I now have comprehensive data. Here is the complete answer:
Meromelia in Neonates — Causes
Definition
Meromelia (Greek: meros = part, melos = limb) refers to partial absence of one or more limbs. It is a type of limb reduction defect that includes:
- Hemimelia — absence of a specific bone segment (e.g., absent fibula)
- Phocomelia — hands/feet attached directly to the trunk by small, irregularly shaped bones ("seal limbs")
- Micromelia — all segments present but abnormally short (sometimes classified separately)
It differs from amelia (complete absence of a limb). Major limb defects occur in approximately 1 in 500 neonates.
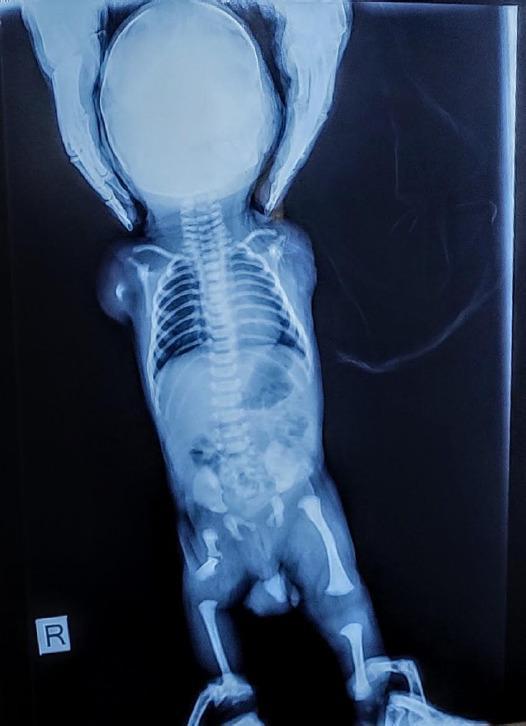
Critical Period
The critical period for limb development is 24–36 days after fertilization (approximately 5th–6th week of gestation). Any insult during this window can disrupt limb formation. Insults early in the critical period tend to produce severe defects (amelia); insults later cause partial absence (meromelia).
Causes
1. Teratogenic (Drug/Chemical) Causes
| Teratogen | Mechanism | Features |
|---|---|---|
| Thalidomide | Inhibits early blood vessel formation in limb buds; also disrupts FGF and angiogenesis | Classic cause; ~12,000 neonates affected 1957–1962; phocomelia, absent/short long bones; associated with anotia/microtia, cardiac defects, intestinal atresia |
| Warfarin | Disrupts vitamin K-dependent bone proteins | Stippled epiphyses, limb hypoplasia |
| Phenytoin | Disrupts folate metabolism and cell proliferation | Digit/limb hypoplasia (fetal hydantoin syndrome) |
| Valproic acid | Folic acid antagonism | Limb reduction, neural tube defects |
| Cocaine | Vascular disruption → ischemia | Limb reduction defects, other defects |
| Misoprostol | Vascular disruption | Möbius sequence, transverse limb defects |
| Alcohol | Multifactorial disruption | Part of fetal alcohol spectrum |
Thalidomide is the classic cause. The sensitive period was 20–36 days after fertilization. Currently still used for leprosy, multiple myeloma, and autoimmune diseases — absolutely contraindicated in women of childbearing age. Teratogen-induced meromelia is usually bilateral and symmetric.
2. Genetic / Chromosomal Causes
Chromosomal:
- Trisomy 18 (Edwards syndrome) — associated with various limb malformations
- Other chromosomal aneuploidies
Mutant genes / Monogenic causes:
- Mutations in HOX genes (HOXA/HOXD family) — master regulators of limb patterning along the proximal-distal and anterior-posterior axes
- BMP (bone morphogenetic proteins) pathway mutations
- SHH (sonic hedgehog) mutations — anterior-posterior patterning
- WNT7 mutations — dorsal-ventral limb patterning
- EN1 mutations
- TBX5 mutations (chromosome 12q24.1) → Holt-Oram syndrome (upper limb abnormalities + congenital heart defects)
- HOXA13 mutations → hand-foot-genital syndrome
- HOXD13 mutations → synpolydactyly
3. Vascular Disruption / Ischemic Causes
Disruption of blood supply during limb development leads to limb reduction defects via ischemia and tissue loss. This is a key non-teratogenic, non-genetic mechanism. It explains some sporadic cases and is implicated in cocaine-associated meromelia.
4. Amniotic Band Syndrome (ABS)
- Occurs in 1:1,200 to 1:15,000 live births
- Fibrous bands of amnion encircle and constrict fetal limbs
- Second or third trimester disruption → constriction rings, lymphedema, and limb amputation (producing transverse meromelia)
- First trimester disruption → craniofacial and visceral defects
- Most commonly affects hands/feet (90%), umbilical cord (30%), abdomen (20%)
- Results in: limb/digit amputation, constriction rings, acrosyndactyly (distal digit fusion)
- ABS-related defects are typically asymmetric and random (unlike teratogen-induced defects)
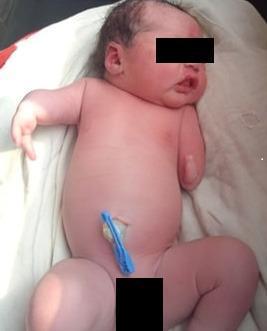
5. Mechanical / Intrauterine Factors
- Oligohydramnios — reduced amniotic fluid commonly associated with limb deformations (compression effects)
- Intrauterine constraint and mechanical pressure during the critical developmental window
6. Multifactorial / Unknown
- A combination of genetic predisposition and environmental triggers
- Most cases of thyroid dysgenesis and limb defects occurring sporadically may have polygenic/epigenetic components
- Many cases remain idiopathic despite full workup
Summary Table
| Category | Examples | Symmetry |
|---|---|---|
| Teratogens | Thalidomide, cocaine, misoprostol, warfarin, phenytoin, valproate | Usually bilateral/symmetric |
| Genetic/Chromosomal | HOX, BMP, SHH mutations; Trisomy 18; Holt-Oram | Variable |
| Vascular disruption | Ischemia in utero | Variable |
| Amniotic band syndrome | Band constriction/amputation | Asymmetric/random |
| Mechanical | Oligohydramnios, compression | Variable |
Sources:
- The Developing Human: Clinically Oriented Embryology — Ch. 16 (Birth Defects of Limbs), Ch. 20 (Teratology)
- Langman's Medical Embryology — Ch. 12 (Limb Defects)
- Creasy & Resnik's Maternal-Fetal Medicine — Ch. 26 (Amniotic Band Syndrome)
Sickle cell anemia mother neonate workup
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Searching PubMed
sickle cell disease neonate newborn screening management
Reading Article
PMID: 38888156
I now have sufficient data to compose a comprehensive answer. Here it is:
Neonate Born to a Mother with Sickle Cell Anemia — Workup
Context: Why the Neonate Needs a Workup
When the mother has sickle cell disease (SCD), the neonate is at risk of:
- Inheriting SCD or sickle cell trait (depending on the father's hemoglobin genotype)
- Neonatal complications related to maternal SCD during pregnancy (growth restriction, prematurity, anemia)
- Early-onset complications of SCD itself if the infant is affected
The key principle: SCD is not clinically apparent at birth because fetal hemoglobin (HbF, α2γ2) predominates and does not sickle. Symptoms emerge only as HbF is replaced by HbS over the first 6–12 months of life. This is exactly why newborn screening is critical — it identifies affected infants before symptoms begin.
Step 1 — Newborn Screening (Universal + Targeted)
Hemoglobin Electrophoresis (Gold Standard)
- The initial and definitive diagnostic test for SCD in the neonatal period
- Performed via heel-stick dried blood spot (filter paper card) or cord blood
- Identifies the specific hemoglobin pattern present
Neonatal Hemoglobin Electrophoresis Patterns:
| Pattern Reported | Interpretation |
|---|---|
| FA | Normal (HbF + HbA) — not affected |
| FAS | Sickle cell trait (HbF + HbA + HbS) — carrier |
| FS | HbSS disease (sickle cell anemia) — most severe |
| FSC | HbSC disease |
| FSA | HbSβ+ thalassemia (milder) |
| FS (no A) | HbSβ0 thalassemia or HbSS (equally severe to SS) |
Important pitfall: The Sickledex (solubility test) and sickle preparation tests can give false-negative results in neonates because the high percentage of HbF dilutes HbS below the detection threshold. These tests should not be used for neonatal screening. Hemoglobin electrophoresis or HPLC (high-performance liquid chromatography) is required.
- In the US, newborn screening for SCD is universal and mandatory in all 50 states
- Screening is ideally done at 2–4 days of life for term infants
HPLC (High-Performance Liquid Chromatography)
- Increasingly used alongside or instead of electrophoresis in newborn screening programs
- Quantifies each hemoglobin fraction precisely and identifies variants
Confirmatory Testing
- Any positive screen must be confirmed with a second independent sample (serum or repeat filter card) using a different method (e.g., electrophoresis at alkaline + acid pH, or HPLC + IEF)
- Confirmation should occur by 4–8 weeks of age
Step 2 — Parental Genotype Assessment
Since the mother is known to have SCD (HbSS):
- Father's hemoglobin status must be determined to assess the neonate's risk
- Father's genotype determines whether the baby can have:
- HbSS (if father carries HbS)
- HbSC (if father carries HbC)
- HbSβ-thalassemia (if father carries β-thal mutation)
- HbAS trait only (if father is HbAA or HbAS)
Ideally, father's hemoglobin electrophoresis is obtained prenatally or at delivery.
Step 3 — Baseline Laboratory Workup (if infant is screen-positive)
Once an infant is identified as having SCD on screening, baseline labs are collected — these values are critical for future comparison during illness:
| Test | Purpose |
|---|---|
| CBC with differential | Baseline hemoglobin, WBC, platelets |
| Reticulocyte count | Baseline erythropoietic activity |
| Hemoglobin electrophoresis (% HbF) | Quantify HbF — protective against sickling |
| Fractionated (direct/indirect) bilirubin | Baseline; hyperbilirubinemia common |
| Liver function tests (ALT, AST) | Baseline hepatic function |
| BUN / Creatinine | Baseline renal function |
| Urinalysis | Baseline renal screen |
In sickle cell disease, typical reticulocyte count is 3–4× the upper limit of normal due to compensated hemolysis. A reticulocyte count ≤3% of normal may signal aplastic crisis; >12% with nucleated RBCs suggests rapid hemolysis.
Step 4 — Imaging and Additional Studies
For a newly diagnosed asymptomatic neonate, imaging is not routinely required at birth. However:
- Transcranial Doppler (TCD) ultrasound: Recommended starting at 2 years of age in HbSS and HbSβ0-thalassemia patients to screen for stroke risk (velocity >200 cm/sec indicates high risk requiring transfusion)
- Echocardiogram: Not routine at birth, but relevant if cardiac symptoms emerge
- Ophthalmology: Screening for retinal disease starts in older children
Step 5 — Preventive Interventions (Initiated in Infancy)
Penicillin Prophylaxis (Critical)
Functional asplenia begins in infancy in HbSS disease (splenic autoinfarction), dramatically increasing risk for overwhelming pneumococcal sepsis — the leading cause of death in young children with SCD.
- Start: As soon as diagnosis is confirmed — by 2 months of age
- Dose (Penicillin V potassium):
- Age 2 months to <3 years: 125 mg PO BID
- Age 3–5 years: 250 mg PO BID
- Can be discontinued after age 5 if the child received all recommended pneumococcal vaccinations and has had no invasive pneumococcal infection
- Covers encapsulated organisms (especially S. pneumoniae)
Immunizations
Strictly follow recommended schedule with emphasis on:
- Pneumococcal vaccines: PCV15/PCV20 (primary series) + PPSV23 after age 2
- Meningococcal vaccines
- Haemophilus influenzae type b (Hib) vaccine
- Annual influenza vaccine
Folic Acid Supplementation
- Ongoing hemolysis increases folate demand; supplementation is standard
Step 6 — Hydroxyurea Considerations
- Hydroxyurea (HU) is now recommended for all children with HbSS or HbSβ0-thalassemia starting at 9 months of age (regardless of clinical severity), per NHLBI guidelines
- Mechanism: induces HbF production → reduces sickling, hemolysis, pain crises, ACS, and mortality
- Not typically initiated in the neonatal period but should be planned from diagnosis
Summary Workup Table
| When | What |
|---|---|
| Birth (days 2–4) | Universal newborn screen: hemoglobin electrophoresis/HPLC on dried blood spot |
| At diagnosis confirmation (by 4–8 wks) | CBC, reticulocyte count, Hb electrophoresis (% HbF), LFTs, BUN/Cr, UA, fractionated bilirubin |
| Parental testing | Father's hemoglobin electrophoresis (ideally prenatal) |
| By 2 months | Start penicillin V prophylaxis |
| At 2 months onward | Pneumococcal, meningococcal, Hib vaccinations per schedule |
| By 9 months | Consider initiating hydroxyurea |
| From age 2 years | Annual transcranial Doppler for stroke screening |
Sources:
- The Harriet Lane Handbook, 23rd Edition — Ch. 14 (Hematology); Ch. Formulary (Penicillin V)
- Rosen's Emergency Medicine, Ch. 109 (Sickle Cell Disease)
- Henry's Clinical Diagnosis and Management by Laboratory Methods — Ch. 38
- Creasy & Resnik's Maternal-Fetal Medicine, Ch. 55
- Mosca A, et al. "Screening for sickle cell disease: focus on newborn investigations." Clin Chem Lab Med 2024 [PMID: 38888156]
Understanding x rays in neonates
Finding Sources
Finding Sources
Finding Sources
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
Generate a comprehensive PDF guide to neonatal chest X-ray interpretation
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.