Pnh
Reading File
Finding Sources
Searching PubMed
"paroxysmal nocturnal hemoglobinuria"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Here is a comprehensive overview of Paroxysmal Nocturnal Hemoglobinuria (PNH):
Paroxysmal Nocturnal Hemoglobinuria (PNH)
Definition
PNH is a rare, acquired clonal blood disorder caused by a somatic mutation in the PIG-A gene (on the X chromosome) in a hematopoietic stem cell. This gene encodes an enzyme essential for synthesis of GPI (glycosylphosphatidylinositol) anchors, which tether numerous proteins to the cell membrane. Loss of GPI anchors means GPI-linked complement regulatory proteins are absent from the surface of red cells, white cells, and platelets - rendering them vulnerable to complement-mediated destruction. - Goldman-Cecil Medicine, p. 1692
Pathophysiology
- The PIG-A mutation occurs in a single hematopoietic stem cell; all clonal progeny (RBCs, WBCs, platelets) are affected
- X-linked location of PIG-A means a single mutation on the active X chromosome suffices (lyonization)
- ~150 different PIG-A mutations have been identified
- The GPI-anchored proteins lost include:
- CD59 (protectin / MIRL - membrane inhibitor of reactive lysis) - the most important; inhibits C3 convertase and prevents C9 polymerization (MAC formation)
- CD55 (DAF - decay-accelerating factor) - antagonizes C3/C5 convertase complexes
- C8-binding protein (homologous restriction factor)
- Also: CD14, CD16a, CD24, CD58, acetylcholinesterase, leukocyte alkaline phosphatase
- Without CD59 and CD55, the membrane attack complex (C5b-9) assembles unopposed on cell surfaces → intravascular hemolysis
Three types of PNH RBCs (based on sensitivity to complement lysis):
| Type | GPI Deficiency | Sensitivity to Lysis |
|---|---|---|
| Type I | Normal | Normal |
| Type II | Partial | 3-5x normal |
| Type III | Complete (no GPI) | 15-25x normal |
- Henry's Clinical Diagnosis and Management, p. 693; Robbins & Kumar Basic Pathology, p. 606
Why PNH Clones Expand
Most normal individuals have a small number of PIG-A-mutant bone marrow cells that remain harmless. They are thought to expand (producing clinical PNH) only when they gain a selective advantage - most commonly in the setting of autoimmune attack against GPI-linked antigens (explaining the strong association with aplastic anemia). PNH clones also upregulate anti-apoptotic genes that make them resistant to immune attack. - Robbins, Cotran & Kumar Pathologic Basis of Disease
Epidemiology
- Incidence: ~2-5 per million/year; prevalence ~1 per million
- Age: most common between 10-50 years (mean ~34, median ~40 years)
- No sex preference (F:M ratio ~1:1)
- No family history typical (acquired, not inherited)
- Median survival after diagnosis: ~20 years
- Goldman-Cecil Medicine, p. 1692
Clinical Features
| Feature | Details |
|---|---|
| Hemoglobinuria | Dark/brown urine on waking - but classic in only 25%; most have chronic hemolysis without dramatic hemoglobinuria |
| Hemosiderinuria | Almost always present - leads to chronic urinary iron loss and iron deficiency |
| Anemia | Normocytic (usually mild-moderate), but can become microcytic/hypochromic due to iron deficiency; often DAT-negative |
| Thrombosis | Leading cause of death; ~40% of patients; affects unusual venous sites (hepatic = Budd-Chiari, portal, cerebral, mesenteric veins) in 85% of thrombotic events |
| Abdominal pain | ~1/3 of patients; from NO scavenging by free hemoglobin |
| Dysphagia | Smooth muscle dystonias due to NO depletion |
| Erectile dysfunction | Same mechanism (NO scavenging) |
| Pancytopenia | Neutropenia in ~3/5, thrombocytopenia in ~2/3 at some time |
| Aplastic anemia | ~1/3 evolve into; 50-60% of aplastic anemia patients have subclinical PNH clones |
| AML transformation | Rare |
- Goldman-Cecil Medicine, p. 1692; Henry's Clinical Diagnosis, p. 693
Triggers of hemolytic crises: infection, surgery, transfusion, contrast dyes, severe exercise, acidosis, hypoxia, fever.
Diagnosis
Suspect PNH when:
- Acquired, Coombs-negative (DAT-negative) intravascular hemolytic anemia
- Especially if concurrent iron deficiency (from hemosiderinuria)
- Unusual site thrombosis (Budd-Chiari, portal vein)
Gold standard: Flow cytometry demonstrating GPI-deficient cells
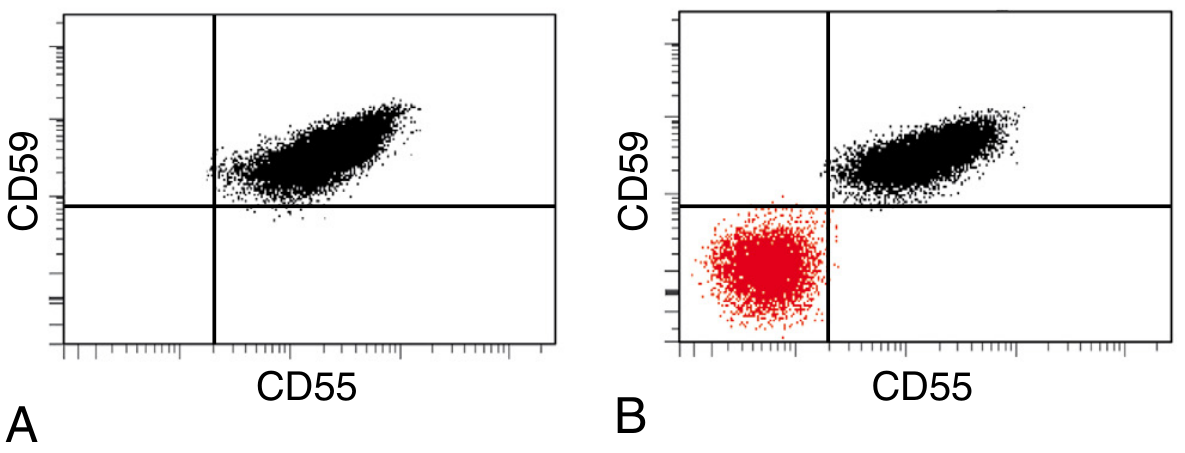
Flow cytometry: Panel A (normal) - all RBCs are CD55+/CD59+. Panel B (PNH patient) - large red cluster in the lower-left quadrant represents the GPI-deficient PNH clone lacking both CD55 and CD59. - Robbins, Cotran & Kumar Pathologic Basis of Disease
- Test both neutrophils/monocytes (CD24, CD57, CD14) and RBCs (CD59)
- FLAER (fluorescent aerolysin variant) - binds directly to the GPI anchor on leukocytes; most reliable reagent for PNH diagnosis
- Clone size correlates with degree of hemolysis
- Old tests (Ham's acidified serum test, sucrose hemolysis test) are obsolete
Treatment
Complement Inhibitors (First-Line where available)
| Drug | Mechanism | Notes |
|---|---|---|
| Eculizumab (anti-C5) | Humanized mAb blocking C5 cleavage, prevents MAC formation | Introduced 2007; abolishes/reduces transfusion requirement; reduces thrombosis; but causes iatrogenic extravascular hemolysis (PNH RBCs now opsonized by C3, Coombs-positive) |
| Ravulizumab (anti-C5) | Long-acting anti-C5 | Less frequent dosing than eculizumab |
| Pegcetacoplan (anti-C3) | Proximal complement inhibitor (C3 level) | Prevents both intravascular AND extravascular hemolysis; more complete anemia correction; risk of more severe breakthrough hemolysis if blockade is incomplete |
- Meningococcal vaccination mandatory before starting anti-C5 therapy (risk of Neisseria meningitidis)
Supportive Treatment
- Folic acid supplements (at least 3 mg/day) - mandatory
- Iron supplementation - check serum iron periodically (lost in urine)
- Transfusions (washed/filtered RBCs) - as needed
- Anticoagulation - for all patients with prior thrombosis or thrombophilic risk factors
- Thrombolysis (tPA) - for thrombotic complications not otherwise resolving
- Glucocorticoids: NOT indicated long-term (no benefit, significant side effects); short course only if inflammatory exacerbation
Bone Marrow Transplantation (BMT)
-
Only curative option
-
Consider for: young patients with severe PNH, PNH-aplastic anemia syndrome (AA), HLA-identical sibling available
-
Anti-C5 therapy has no effect on underlying bone marrow failure (BMF)
-
Immunosuppression (ATG + cyclosporine) is an alternative for PNH-AA patients
-
Harrison's Principles of Internal Medicine 22E, p. 849-850; Goldman-Cecil Medicine, p. 1692
PNH Categories (WHO)
- Classic PNH - hemolysis dominant; large clone
- PNH in the setting of another bone marrow disorder (AA, MDS) - cytopenias predominant; clone often small
- Subclinical PNH - PIG-A mutant cells detectable but no hemolysis or thrombosis
Key Associations to Remember
- PNH + Aplastic Anemia: very common overlap (50-60% of AA patients have subclinical PNH clones)
- Budd-Chiari syndrome in a young patient = think PNH
- DAT-negative hemolysis + iron deficiency = think PNH
- Free hemoglobin scavenges NO → smooth muscle dystonia (abdominal pain, dysphagia, erectile dysfunction)
- CD59 deficiency on platelets → phosphatidylserine externalization → prothrombinase complex formation → thrombosis
This is a shared conversation. Sign in to Orris to start your own chat.