Leverticitam
Checking Date & Time
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I have sufficient information from multiple authoritative textbooks. Here is a comprehensive summary of Levetiracetam (which it looks like you were searching for — noting the alternate spelling "Leverticitam"):
Levetiracetam (Keppra)
Trade names: Keppra, Keppra XR, Elepsia XR, Roweepra, Spritam
Drug Class & Overview
Levetiracetam is a broad-spectrum antiseizure (anticonvulsant) agent and one of the most widely prescribed drugs for epilepsy. It was originally developed as a second-generation nootropic (memory-enhancing) agent and proved to be a potent anticonvulsant. It was FDA-approved in 1999 as adjunctive therapy for refractory partial seizures.
Its popularity stems from:
- Broad therapeutic window
- Favorable pharmacokinetic profile
- Minimal drug–drug interactions
- No hepatic metabolism
Mechanism of Action
Levetiracetam binds selectively to SV2A — a ubiquitous synaptic vesicle integral membrane protein that functions as a positive effector of synaptic vesicle exocytosis. The drug accesses recycling synaptic vesicles by vesicular endocytosis; binding to SV2A reduces glutamate release during high-frequency neuronal activity.
Additional actions include:
- GABAergic effects
- Anti-kindling effects
- Reduction of voltage-dependent potassium currents
It is not active in the standard MES (maximal electroshock) or PTZ (pentylenetetrazole) acute seizure models, but is potently active in chronic kindling models and genetic epilepsy models.
Clinical Indications
| Indication | Notes |
|---|---|
| Focal (partial) seizures | Adults & children (adjunctive or monotherapy) |
| Primary generalized tonic-clonic seizures | Age ≥6 years |
| Myoclonic seizures of juvenile myoclonic epilepsy (JME) | Age ≥12 years |
| Migraine prophylaxis (children) | Evidence from RCT |
| Status epilepticus | IV formulation available |
Pharmacokinetics
| Parameter | Value |
|---|---|
| Bioavailability | >95% (unaffected by food) |
| Time to peak plasma conc. | ~1.3 hours |
| Protein binding | <10% |
| Volume of distribution | ~0.6 L/kg |
| Half-life | 6–8 hours (longer in elderly) |
| Metabolism | ~24–34% hydrolysis in blood (not liver) → inactive metabolite (2-pyrrolidone-N-butyric acid) |
| Excretion | ~66% unchanged in urine |
| Drug interactions | Minimal (no CYP450 involvement) |
| Hepatic impairment | No dose adjustment needed |
| Renal impairment | Dose reduction required |
| Therapeutic range | 12–46 µg/mL (70–270 µmol/L) |
Clearance increases in the third trimester of pregnancy — monitoring and dose adjustment may be needed.
Dosing
Adults (immediate-release):
- Start: 500–1000 mg/day in 2 divided doses
- Increase by 1000 mg/day every 2–4 weeks
- Maximum: 3000 mg/day
Pediatric (partial seizures, immediate-release/IV):
- Infants 1–5 months: Start 7 mg/kg/dose BID → target 21 mg/kg/dose BID
- Older children: Weight-based dosing with gradual titration
Formulations: Immediate-release tablets (250, 500, 750, 1000 mg), extended-release tablets, oral solution (100 mg/mL), IV injection (100 mg/mL)
Adverse Effects
Common:
- Somnolence (~15% in adults, ~23% in children)
- Asthenia/fatigue
- Dizziness, ataxia
- Nasopharyngitis/infection
Behavioral/Psychiatric (important):
- Irritability, aggression, agitation, anger
- Anxiety, depression, emotional lability
- Hostility, apathy, depersonalization
- In children, behavioral problems occur in 37.6% (vs. 18.6% with placebo)
- Behavioral effects tend to appear early (often within the first month)
- Suicidality: Increased risk, as with all antiseizure medications (0.5% vs. 0% with placebo in trials)
Overdose: Somnolence, agitation, aggression, respiratory depression, coma
Drug Interactions
Levetiracetam has a low interaction potential due to renal elimination and non-CYP metabolism:
- Does not significantly affect carbamazepine, lamotrigine, valproate, gabapentin, or topiramate levels
- Carbamazepine may decrease levetiracetam levels (clinical significance variable)
- Oxcarbazepine may modestly decrease levetiracetam levels
- Combination with carbamazepine or topiramate may cause pharmacodynamic (not pharmacokinetic) adverse effects
Pregnancy & Lactation
- Pregnancy: Classified as Category C. Associated with a lower risk of major congenital anomalies than most other antiepileptic medications studied. Fetal loss and teratogenicity seen in animals but not firmly established in humans. Clearance increases in the third trimester.
- Lactation: Classified as "probably safe," but is excreted in breast milk — use only if benefits clearly outweigh risks.
Key Distinguishing Features
- No hepatic metabolism → safe with liver disease; dose reduce with renal disease
- Minimal drug–drug interactions — favorable in polypharmacy patients
- Available IV → useful in status epilepticus and patients unable to take oral medications
- Psychiatric side effects are clinically significant — monitor patients, especially early in treatment
Sources: Katzung's Basic and Clinical Pharmacology 16th ed., Kaplan & Sadock's Comprehensive Textbook of Psychiatry, Tietz Textbook of Laboratory Medicine 7th ed., The Harriet Lane Handbook 23rd ed., Creasy & Resnik's Maternal-Fetal Medicine
Phenytoin
Checking Date & Time
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
I now have comprehensive content from multiple authoritative sources. Here is a complete reference summary:
Phenytoin (Dilantin)
Trade names: Dilantin, Phenytek, generics | Prodrug: Fosphenytoin (Cerebyx)
Drug Class & Overview
Phenytoin is the oldest non-sedating antiseizure medication, first identified in 1938. Chemically, it is the 5,5-diphenyl-substituted analog of hydantoin (diphenylhydantoin — DPH), a five-membered ring compound structurally similar to but distinct from barbiturates. While historically a first-line agent, it is no longer considered first-line for chronic therapy due to its adverse effect profile, nonlinear kinetics, and extensive drug–drug interactions.
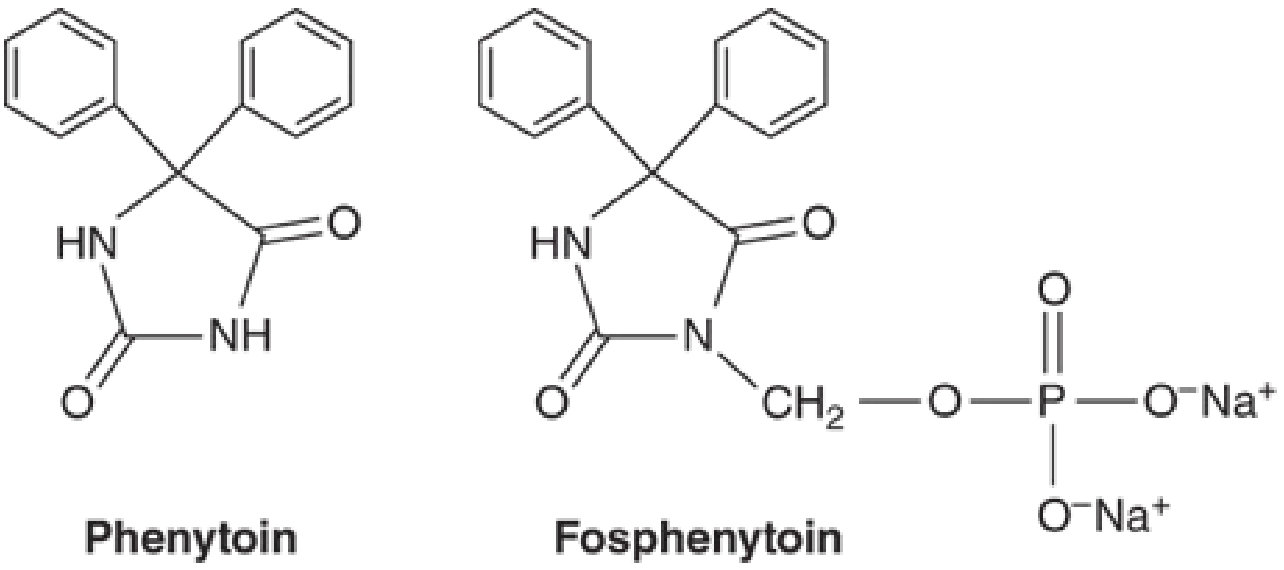
Mechanism of Action
Phenytoin is a voltage-gated sodium channel blocker. It limits repetitive firing of action potentials by slowing the rate of recovery of Na⁺ channels from inactivation. This effect is:
- Voltage-dependent: greater effect when the membrane is depolarized
- Use-dependent: more effect on neurons firing at high frequency
At therapeutic concentrations, the effect on Na⁺ channels is selective, with no changes in spontaneous activity or GABA/glutamate responses. At 5–10× therapeutic concentrations, broader CNS depression occurs, underlying toxicity.
Clinical Indications
| Indication | Notes |
|---|---|
| Focal (partial) onset seizures | First described use |
| Generalized tonic-clonic seizures | Including secondarily generalized |
| Status epilepticus (acute) | IV/fosphenytoin preferred |
| Cardiac arrhythmias | Historically used (same therapeutic range) |
| NOT effective | Absence seizures |
| May worsen | Absence epilepsy, juvenile myoclonic epilepsy (JME), Dravet syndrome |
Pharmacokinetics
| Parameter | Detail |
|---|---|
| Absorption | Nearly complete but formulation-dependent; peak at 3–12 hours |
| Protein binding | ~90–95% to albumin — highly prone to displacement |
| Metabolism | Hepatic CYP2C9/CYP2C19 hydroxylation → inactive 5-(p-hydroxyphenyl)-5-phenylhydantoin, excreted as glucuronide |
| Kinetics | Zero-order (nonlinear/saturation) kinetics within the therapeutic range — small dose increases → large concentration jumps |
| Half-life | Variable (7–42 hours), dose-dependent |
| Excretion | Renal (as glucuronide metabolite) |
| Free fraction | ~10% (but highly variable: 3–37% in individuals) |
⚠️ Critical: Phenytoin follows Michaelis-Menten (saturation) kinetics, not first-order. Once metabolism is saturated (~5 µg/mL), small dose increments cause disproportionately large rises in plasma concentration → narrow margin between efficacy and toxicity.
Therapeutic Drug Monitoring
| Level | Target |
|---|---|
| Total phenytoin | 10–20 µg/mL (40–79 µmol/L) |
| Free phenytoin | 1–2 µg/mL (1–8 µmol/L) |
| Toxic (nystagmus/ataxia) | >20 µg/mL |
| Paradoxical seizure activity | >35 µg/mL |
Monitor free phenytoin in hypoalbuminemia, renal insufficiency, elderly, neonates, and drug-displacement states.Trough sampling (before next dose) is standard for routine monitoring. Peak (4–5 hours post-dose) is used for suspected toxicity.
Dosing
Adults:
- Start: 300 mg/day (divided or extended-release once daily)
- Increase in increments of no more than 25–30 mg/day at a time — due to saturation kinetics
- ⚠️ Common error: jumping from 300 → 400 mg/day causes toxicity
IV Loading (status epilepticus):
- Use fosphenytoin IV (preferred over phenytoin IV) — better solubility, less risk of purple glove syndrome
- Max infusion rate: ≤50 mg/min (phenytoin); slower in neonates (0.5 mg/kg/min), infants/children (1 mg/kg/min)
- Risk of cardiovascular collapse with rapid IV infusion
Children:
- 5 mg/kg/day initial; adjust after steady-state levels obtained
- Extended-release: once or twice daily; chewable tablets/suspension: TID
CYP2C9 phenotype adjustments:
- Intermediate metabolizer: 25% dose reduction + TDM
- Poor metabolizer: 50% dose reduction + TDM
- HLA-B*15:02 or CYP2C9*3 carriers: consider avoiding phenytoin
Formulations
- Oral: Extended-release capsules (sodium salt, preferred for once-daily dosing), immediate-release suspension, chewable tablets
- IV: Phenytoin sodium injection (contains propylene glycol, pH 12 — poorly soluble, irritating)
- Fosphenytoin (Cerebyx): water-soluble prodrug → rapidly converted to phenytoin in plasma; preferred for IV and IM use
Adverse Effects
Dose-Related (Concentration-Dependent)
| Plasma Level | Manifestations |
|---|---|
| Therapeutic (10–20 µg/mL) | Nystagmus, loss of smooth pursuit — not an indication to reduce dose |
| 20–30 µg/mL | Diplopia, ataxia — most common effects requiring dose reduction |
| >30 µg/mL | Sedation, slurred speech |
| >35 µg/mL | Paradoxical seizure activity |
Chronic/Long-Term Effects
- Gingival hyperplasia — occurs to some degree in most patients (not concentration-related)
- Hirsutism — especially distressing in women
- Coarsening of facial features
- Peripheral neuropathy — diminished DTRs in lower extremities
- Osteomalacia — abnormal vitamin D metabolism
- Folate deficiency / megaloblastic anemia
Idiosyncratic (Rare)
- Skin rash / hypersensitivity — can progress to severe exfoliative dermatitis (Stevens-Johnson syndrome)
- Fever, lymphadenopathy (distinguish from malignant lymphoma)
- Agranulocytosis (rare; with fever and rash)
IV Administration
- Purple glove syndrome — purplish-black discoloration, edema, pain distal to injection site
- Cardiovascular collapse with rapid infusion (hypotension, bradycardia, arrhythmia)
Drug Interactions
Phenytoin has extensive and complex drug interactions due to its CYP2C9/2C19 metabolism, high protein binding, and its own potent enzyme-inducing properties.
Drugs that DECREASE phenytoin levels (enzyme inducers):
- Carbamazepine, phenobarbital, alcohol (chronic use)
Drugs that INCREASE phenytoin levels (enzyme inhibitors):
- Isoniazid, chloramphenicol, cimetidine, disulfiram, dicumarol
Drugs that displace phenytoin from albumin (reduce total but not free level):
- Valproate (also inhibits metabolism — increases free phenytoin significantly), salicylates, sulfonylureas, phenylbutazone, sulfisoxazole
Drugs AFFECTED by phenytoin (phenytoin is a potent inducer of CYP1A2, CYP2C, CYP3A4, UGT):
- Reduces levels of: warfarin, oral contraceptives, corticosteroids, cyclosporine, theophylline, neuromuscular blockers, many antiretrovirals
- Reduces T3/T4 (without clinical hypothyroidism)
- May cause hyperammonemia with valproic acid
Pregnancy & Special Populations
- Teratogen: Associated with fetal hydantoin syndrome (craniofacial abnormalities, digital hypoplasia, growth restriction, cognitive effects)
- Protein binding: Increased free fraction in neonates, elderly, hypoalbuminemia (liver disease, nephrotic syndrome) — toxicity at "normal" total levels
- Ideal body weight should be used for dosage calculations
- Steady state typically achieved after 5–10 days of continuous dosing
Sources: Katzung's Basic and Clinical Pharmacology 16th ed., Goodman & Gilman's Pharmacological Basis of Therapeutics, Tietz Textbook of Laboratory Medicine 7th ed., The Harriet Lane Handbook 23rd ed.
AEDs with comparison
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive comparative data from multiple authoritative sources. Let me compile the full AED comparison.
Antiepileptic Drugs (AEDs) — Comprehensive Comparison
Also called Antiseizure Medications (ASMs). Goal of treatment: complete seizure freedom without adverse effects. ~65% of patients achieve remission on medication; ~45–50% respond to the first agent chosen.
1. Mechanism of Action — Classification
| Class | Drugs | Mechanism |
|---|---|---|
| Na⁺ channel blockers | Phenytoin, Carbamazepine, Oxcarbazepine, Eslicarbazepine, Lamotrigine, Lacosamide, Zonisamide | Prolong Na⁺ channel inactivation → reduce high-frequency firing |
| Na⁺ channel blockers (slow inactivation) | Lacosamide | Unique slow-inactivation kinetics at Na⁺ channels |
| Ca²⁺ channel blockers (T-type) | Ethosuximide, Valproate | Block thalamic T-type Ca²⁺ channels → suppress absence seizures |
| GABA-A enhancers (benzodiazepine site) | Clonazepam, Clobazam, Diazepam, Lorazepam | Enhance Cl⁻ influx via GABA-A receptor |
| GABA-A enhancers (barbiturate site) | Phenobarbital, Primidone | Prolong Cl⁻ channel opening |
| GABA reuptake inhibitor | Tiagabine | Blocks GAT-1 transporter → increases synaptic GABA |
| GABA transaminase inhibitor | Vigabatrin | Irreversibly inhibits GABA-T → increases GABA |
| SV2A ligands | Levetiracetam, Brivaracetam | Bind synaptic vesicle protein SV2A → reduce glutamate release |
| AMPA receptor antagonist | Perampanel | Blocks AMPA glutamate receptors |
| Multiple / broad | Valproate, Topiramate, Zonisamide | Multiple mechanisms (Na⁺/Ca²⁺ channels + GABA potentiation + glutamate blockade) |
| Carbonic anhydrase inhibitor | Topiramate, Zonisamide, Acetazolamide | Inhibit carbonic anhydrase |
| HCN channel blocker | Gabapentin, Pregabalin | Bind α2δ subunit of voltage-gated Ca²⁺ channels → reduce excitatory neurotransmitter release |
| mTOR inhibitor | Everolimus | Reduces seizures in tuberous sclerosis |
| Cannabinoid | Cannabidiol (Epidiolex) | Multiple; effective in Dravet syndrome & Lennox-Gastaut |
2. Seizure Type Efficacy — Comparison Table
| Drug | Focal | GTC | Absence | Myoclonic | LGS | Infantile Spasms | Special Uses |
|---|---|---|---|---|---|---|---|
| Phenytoin | ✅ | ✅ | ❌ (worsens) | ❌ | ❌ | ❌ | Status epilepticus (IV) |
| Carbamazepine | ✅ | ✅ | ❌ (worsens) | ❌ | ❌ | ❌ | Trigeminal neuralgia, bipolar |
| Oxcarbazepine | ✅ | ✅ | ❌ | ❌ | ❌ | ❌ | Fewer interactions than CBZ |
| Lamotrigine | ✅ | ✅ | ✅ | ⚠️ (may worsen) | ✅ | ❌ | Bipolar depression |
| Valproate | ✅ | ✅ | ✅ | ✅ | ✅ | ❌ | TRUE broad-spectrum; bipolar |
| Ethosuximide | ❌ | ❌ | ✅ | ✅ | ❌ | ❌ | Drug of choice for absence |
| Levetiracetam | ✅ | ✅ | ❌ | ✅ (JME) | ❌ | ❌ | Fewer interactions; IV available |
| Phenobarbital | ✅ | ✅ | ❌ | ❌ | ❌ | ✅ | Neonatal seizures; status |
| Topiramate | ✅ | ✅ | ❌ | ❌ | ✅ | ❌ | Migraine prophylaxis, obesity |
| Lacosamide | ✅ | ❌ | ❌ | ❌ | ❌ | ❌ | Focal seizures; minimal interactions |
| Zonisamide | ✅ | ✅ | ✅ | ✅ | ❌ | ❌ | Broad-spectrum |
| Gabapentin | ✅ | ✅ | ❌ | ❌ | ❌ | ❌ | Neuropathic pain, anxiety |
| Pregabalin | ✅ | ❌ | ❌ | ❌ | ❌ | ❌ | Neuropathic pain, fibromyalgia |
| Clonazepam | ❌ | ❌ | ✅ | ✅ | ❌ | ❌ | Myoclonic, absence; tolerance develops |
| Vigabatrin | ✅ | ❌ | ❌ | ❌ | ❌ | ✅ | Drug of choice in tuberous sclerosis IS |
| Rufinamide | ❌ | ❌ | ❌ | ❌ | ✅ | ❌ | LGS adjunct |
| Clobazam | ✅ | ❌ | ❌ | ❌ | ✅ | ❌ | LGS; Dravet adjunct |
| Cannabidiol | ❌ | ❌ | ❌ | ❌ | ✅ | ❌ | Dravet syndrome, LGS |
| Perampanel | ✅ | ✅ | ❌ | ❌ | ❌ | ❌ | Adjunctive |
| Brivaracetam | ✅ | ❌ | ❌ | ❌ | ❌ | ❌ | Focal; fewer psych side effects vs LEV |
LGS = Lennox-Gastaut Syndrome | IS = Infantile Spasms | JME = Juvenile Myoclonic Epilepsy | GTC = Generalized Tonic-Clonic
3. Pharmacokinetics Comparison
| Drug | Trade Name | Adult Dose (mg/d) | Peds (mg/kg/d) | Half-life (h) | Therapeutic Level (µg/mL) | Protein Binding |
|---|---|---|---|---|---|---|
| Phenytoin | Dilantin | 300–400 | 4–7 | 12–36 (nonlinear) | 10–20 | ~90–95% |
| Carbamazepine | Tegretol | 600–1,200 | 20–30 | 14–25 (autoinduction) | 4–12 | 75% |
| Oxcarbazepine | Trileptal | 900–2,400 | 10–40 | 1–5 (prodrug; MHD active) | — | 40% (MHD) |
| Eslicarbazepine | Aptiom | 400–1,200 | — | 13–20 | — | — |
| Valproate | Depakote | 1,000–3,000 | 30–60 | 6–15 | 50–100 | ~90% |
| Phenobarbital | Luminal | 90–200 | 3–5 (8 infants) | 40–120 | 15–40 | 50% |
| Lamotrigine | Lamictal | 300–500 | 0.5 | 15–60 (varies by co-meds) | 2–7 | 55% |
| Levetiracetam | Keppra | 500–3,000 | 20–60 | 6–8 | 12–46 | <10% |
| Brivaracetam | Briviact | 50–200 | — | 9 | 0.2–2 | — |
| Topiramate | Topamax | 400 | — | 20–30 | — | 15–25% |
| Lacosamide | Vimpat | 300–400 | 6–8 mg/kg | 12–14 | — | <30% |
| Zonisamide | Zonegran | 100–600 | — | 63 | — | 50% |
| Ethosuximide | Zarontin | 750–1,500 | 20–40 | 20–60 | 50–100 | Minimal |
| Gabapentin | Neurontin | 900–3,600 | — | 5–7 | — | <3% |
| Pregabalin | Lyrica | 150–600 | — | 6 | — | <3% |
| Perampanel | Fycompa | 2–12 | — | 105 | — | 95% |
| Clonazepam | Klonopin | 1.5–20 | 0.01–0.2 | 18–50 | 20–80 ng/mL | 85% |
| Vigabatrin | Sabril | 2,000–3,000 | 100–300 | 5–7 | — | 0% |
4. Adverse Effects — Comparison
| Drug | Key/Unique Adverse Effects |
|---|---|
| Phenytoin | Nystagmus, ataxia, gingival hyperplasia, hirsutism, coarsening facial features, peripheral neuropathy, osteomalacia, megaloblastic anemia, Steven-Johnson syndrome, purple glove syndrome (IV), teratogen (fetal hydantoin syndrome) |
| Carbamazepine | Nausea, diplopia, ataxia, hyponatremia (SIADH), leukopenia, aplastic anemia (rare), Stevens-Johnson (HLA-B*1502), teratogen, enzyme inducer |
| Oxcarbazepine | Hyponatremia (more common than CBZ), dizziness, diplopia, rash; fewer drug interactions than CBZ |
| Valproate | Hepatotoxicity (esp. <2 yrs, polytherapy), pancreatitis, teratogen (neural tube defects, fetal valproate syndrome), tremor, weight gain, hair loss, thrombocytopenia, hyperammonemia |
| Lamotrigine | Rash/Stevens-Johnson (esp. rapid titration or with valproate), dizziness, headache, diplopia; safe in pregnancy relative to others |
| Levetiracetam | Behavioral/psychiatric effects (irritability, aggression, depression — 37% in children), somnolence; minimal drug interactions |
| Phenobarbital | Sedation, cognitive dulling, depression, paradoxical hyperactivity in children, tolerance, dependence, teratogen; long half-life |
| Topiramate | Cognitive slowing/word-finding difficulty, weight loss (useful clinically), kidney stones (carbonic anhydrase), metabolic acidosis, glaucoma, teratogen (cleft lip) |
| Ethosuximide | Nausea, headache, hiccups, behavioral changes; rarely SLE-like syndrome, aplastic anemia |
| Gabapentin/Pregabalin | Sedation, dizziness, weight gain, peripheral edema; pregabalin has abuse potential |
| Lacosamide | Dizziness, nausea, cardiac conduction effects (PR prolongation) |
| Zonisamide | Sedation, kidney stones, metabolic acidosis, weight loss, oligohidrosis/hyperthermia in children |
| Vigabatrin | Irreversible visual field defects (40% of patients) — requires monitoring |
| Perampanel | Aggression, dizziness, somnolence, weight gain |
| Tiagabine | Dizziness, confusion; can precipitate absence-like status in non-epileptic patients |
| Clonazepam | Sedation, hypersalivation, tolerance with long-term use |
5. Drug Interactions — Key Points
| Category | Drugs |
|---|---|
| Strong enzyme INDUCERS (reduce levels of other drugs) | Phenytoin, Carbamazepine, Phenobarbital, Primidone, Rufinamide — reduce OCP, warfarin, cyclosporine, antiretrovirals |
| Enzyme INHIBITOR | Valproate — inhibits CYP2C9, epoxide hydrolase; raises lamotrigine, phenobarbital levels |
| Minimal interactions (renally cleared or non-CYP) | Levetiracetam, Gabapentin, Pregabalin, Lacosamide, Brivaracetam |
| Valproate + Lamotrigine | Valproate doubles lamotrigine half-life → requires slow titration to avoid SJS rash |
| Valproate + Phenytoin | Valproate displaces phenytoin from albumin AND inhibits metabolism → increased free phenytoin toxicity |
6. Special Patient Populations
| Scenario | Preferred Agents | Agents to Avoid |
|---|---|---|
| Women of childbearing age | Lamotrigine, Levetiracetam | Valproate (highest teratogen risk), Phenytoin, Phenobarbital |
| Absence seizures | Ethosuximide (first-line), Valproate, Lamotrigine | Phenytoin, Carbamazepine, Gabapentin (worsen absence) |
| JME (Juvenile Myoclonic Epilepsy) | Valproate, Levetiracetam, Lamotrigine | Carbamazepine, Phenytoin (may worsen) |
| Dravet syndrome | Valproate, Clobazam, Cannabidiol, Stiripentol | Sodium channel blockers (worsen) |
| Infantile spasms | Vigabatrin (tuberous sclerosis), ACTH | — |
| Lennox-Gastaut | Valproate, Lamotrigine, Rufinamide, Cannabidiol, Clobazam | — |
| Status epilepticus (acute) | Lorazepam/Diazepam → Fosphenytoin/Phenobarbital → Levetiracetam IV → Anesthesia | — |
| Elderly / polypharmacy | Levetiracetam, Gabapentin, Lacosamide, Brivaracetam | Enzyme inducers/inhibitors |
| Renal impairment | Valproate, Carbamazepine | Levetiracetam (needs dose reduction), Gabapentin, Pregabalin |
| Liver disease | Levetiracetam, Gabapentin, Lacosamide | Valproate, Phenytoin, Carbamazepine |
| Neonatal seizures | Phenobarbital (first-line), Phenytoin | — |
| Focal epilepsy (newly diagnosed) | Lamotrigine > Levetiracetam, Zonisamide | — |
| Generalized epilepsy (newly diagnosed) | Valproate > Levetiracetam | — |
| Bipolar disorder + epilepsy | Valproate, Lamotrigine, Carbamazepine | — |
| Neuropathic pain + epilepsy | Gabapentin, Pregabalin, Carbamazepine (TN) | — |
| Obesity | Topiramate, Zonisamide (weight loss) | Valproate, Pregabalin (weight gain) |
| Migraine prophylaxis | Topiramate, Valproate | — |
7. Generation Classification
| Generation | Drugs |
|---|---|
| 1st generation | Phenobarbital, Phenytoin, Carbamazepine, Valproate, Ethosuximide, Clonazepam, Primidone |
| 2nd generation | Lamotrigine, Gabapentin, Topiramate, Oxcarbazepine, Levetiracetam, Tiagabine, Zonisamide, Vigabatrin, Felbamate |
| 3rd generation | Lacosamide, Eslicarbazepine, Brivaracetam, Perampanel, Rufinamide, Clobazam, Cannabidiol, Cenobamate |
⚠️ Note: Newer generation ≠ better efficacy. A 30-year longitudinal analysis found tolerability of newer AEDs was no better than older AEDs overall. Choice depends on seizure type, patient-specific factors, and comorbidities.
8. Quick Reference Summary Card
| Drug | Best For | Avoid In | Key Concern |
|---|---|---|---|
| Phenytoin | Focal, GTC, status | Absence, JME, Dravet | Nonlinear kinetics, gingival hyperplasia |
| Carbamazepine | Focal, trigeminal neuralgia | Absence, JME | Hyponatremia, SJS, enzyme inducer |
| Valproate | All seizure types, bipolar | Pregnancy | Hepatotoxicity, teratogen |
| Ethosuximide | Absence ONLY | Focal, GTC | GI side effects |
| Lamotrigine | Broad-spectrum, bipolar | Rapid titration | SJS rash — titrate slowly |
| Levetiracetam | Broad, IV availability | Psychiatric history | Behavioral side effects |
| Phenobarbital | Neonates, status | Adults (sedation) | Sedation, dependence |
| Topiramate | Focal, GTC, migraine, obesity | Cognitive demands | Word-finding difficulty, stones |
| Lacosamide | Focal | Cardiac disease | PR prolongation |
| Gabapentin | Focal, neuropathic pain | Absence | Minimal efficacy, weight gain |
| Vigabatrin | IS (tuberous sclerosis) | — | Irreversible visual field loss |
| Perampanel | Focal, GTC (adjunct) | — | Aggression, very long half-life |
| Cannabidiol | Dravet, LGS | — | Drug interactions via CYP |
| Clonazepam | Absence, myoclonic | Long-term use | Tolerance |
Sources: Katzung's Basic and Clinical Pharmacology 16th ed., Adams and Victor's Principles of Neurology 12th ed., Goldman-Cecil Medicine, Bradley and Daroff's Neurology in Clinical Practice
This is a shared conversation. Sign in to Orris to start your own chat.