Why beta hcg causes nausea and vomitings
Reading File
Finding Sources
Searching PubMed
beta hCG nausea vomiting pregnancy mechanism
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Here is a thorough, textbook-based explanation of why beta-hCG causes nausea and vomiting in pregnancy:
Why Beta-hCG Causes Nausea and Vomiting in Pregnancy
1. Temporal Correlation (the strongest indirect evidence)
Beta-hCG rises sharply after implantation, peaks at 8-12 weeks of gestation, and then declines to a plateau after 18-20 weeks. This timeline mirrors the course of nausea and vomiting of pregnancy (NVP) almost exactly - symptoms begin around weeks 4-6, peak between weeks 10-16, and resolve by ~20 weeks in most women. Conditions that produce abnormally high hCG levels (molar pregnancy, multiple gestation, Down syndrome, choriocarcinoma) are all associated with more severe nausea and vomiting, further supporting causality.
- Creasy & Resnik's Maternal-Fetal Medicine: "The close temporal association of peak hCG levels and nausea and vomiting symptoms and the fact that conditions associated with elevated levels (e.g., female fetal sex, twins, hydatidiform moles) have higher rates of hyperemesis suggest a causal relationship." Of 15 prospective comparative studies published since 1990, 11 showed significantly higher hCG levels in women with hyperemesis.
2. Cross-reactivity with the TSH Receptor - the key molecular mechanism
The alpha (α) subunit of hCG is structurally identical to the α-subunits of TSH, FSH, and LH. Because of this shared homology, hCG can weakly but directly stimulate TSH receptors on thyroid follicular cells. This leads to:
- Increased thyroid hormone (T4/T3) production
- Suppression of TSH (occurs in up to 20% of normal pregnancies in the first trimester)
- A state called Gestational Transient Thyrotoxicosis (GTT) in 2-5% of pregnancies
The resulting mild hyperthyroidism is a well-recognized cause of nausea and vomiting. In severe cases, it progresses to hyperemesis gravidarum - weight loss >5% of pre-pregnancy weight, dehydration, electrolyte disturbances, and ketonuria.
- Creasy & Resnik's: "hCG directly stimulates TSH receptors due to its similar homology to TSH... GTT occurs in 2-5% of pregnancies and it is often accompanied by hCG-induced nausea or vomiting."
- Yamada's Textbook of Gastroenterology: "There is cross-reactivity between the α-subunit of hCG and the thyroid stimulating hormone (TSH) receptor. 'Biochemical thyrotoxicosis'... may be found in up to two-thirds of women with hyperemesis gravidarum."
3. Stimulation of Estrogen Production from the Ovary
hCG stimulates ovarian granulosa/luteal cells (via LH/hCG receptors) to produce estrogen. Elevated estrogen is itself a potent trigger for nausea:
-
Women who experience nausea on estrogen-containing oral contraceptive pills have a higher incidence of NVP.
-
Of 17 studies examining non-thyroid hormones and NVP, only hCG and estrogen showed a consistent association.
-
Yamada's: "hCG... may contribute to the development of NVP and HG by stimulating estrogen production from the ovary."
-
Sleisenger & Fordtran's: "Increased HCG levels during the first trimester may stimulate gastric CCK receptors and trigger symptoms."
4. Stimulation of Gastric CCK Receptors
hCG has been proposed to directly stimulate cholecystokinin (CCK) receptors in the stomach. CCK is a known mediator of nausea and is involved in the feedback signaling between the gut and the brain's vomiting centers (area postrema / chemoreceptor trigger zone).
5. Gastric Dysmotility
hCG (via estrogen and progesterone) contributes to:
- Reduced lower esophageal sphincter (LES) pressure
- Impaired esophageal peristalsis
- Gastric dysrhythmias (electrogastrographic studies show abnormal slow-wave frequency correlating with symptoms)
- Delayed gastric emptying
These motility changes amplify the nausea signals from hormonal stimulation.
6. Other Contributing Factors
| Factor | Role |
|---|---|
| Helicobacter pylori | Found at higher rates in HG; may worsen hormone-induced motility changes |
| Psychological factors | Depression, stress, anxiety can amplify perception of nausea |
| Olfactory hypersensitivity | Thought to be partly estrogen-mediated |
| Genetic predisposition | Higher concordance in monozygotic twins; family history increases risk |
| Evolutionary adaptation | One hypothesis: NVP protects the fetus from potentially teratogenic foods in the first trimester |
Clinical Spectrum
| Condition | Severity |
|---|---|
| Morning sickness (NVP) | Mild to moderate; 50-80% of pregnancies; self-limiting |
| Hyperemesis Gravidarum (HG) | Severe; 0.3-3% of pregnancies; requires hospitalization, IV fluids, antiemetics |
Summary
The main mechanism is a multi-hit hormonal cascade: beta-hCG cross-stimulates TSH receptors (causing transient thyrotoxicosis) AND drives ovarian estrogen production, and both hormones synergistically derange gastric motility and stimulate central/peripheral nausea pathways. The fact that NVP tracks hCG levels so precisely - peaking at weeks 8-12 and resolving as hCG declines - is the most compelling evidence for this relationship.
Sources: Creasy & Resnik's Maternal-Fetal Medicine; Yamada's Textbook of Gastroenterology, 7th ed.; Sleisenger & Fordtran's Gastrointestinal and Liver Disease; Harrison's Principles of Internal Medicine, 22nd ed.
Wt is the no.of standard WHO visits in pregnancy?
Finding Sources
Searching the Web
WHO recommended number antenatal care contacts visits pregnancy 2016 guidelines
The WHO data is confirmed. Here is the complete answer:
WHO Recommended ANC Contacts in Pregnancy
The 2016 WHO ANC Guidelines ("WHO Recommendations on Antenatal Care for a Positive Pregnancy Experience") recommend a minimum of 8 contacts during pregnancy - increased from the previous 4-visit "Focused Antenatal Care" (FANC) model.
The term "contact" is used deliberately instead of "visit" - it implies an active, meaningful connection between the woman and her healthcare provider, not just a routine check-in.
Schedule of the 8 Contacts
| Contact | Gestational Age |
|---|---|
| Contact 1 | Up to 12 weeks (1st trimester) |
| Contact 2 | 20 weeks |
| Contact 3 | 26 weeks |
| Contact 4 | 30 weeks |
| Contact 5 | 34 weeks |
| Contact 6 | 36 weeks |
| Contact 7 | 38 weeks |
| Contact 8 | 40 weeks |
Why Was It Changed from 4 to 8?
- Evidence showed that 8+ contacts reduce perinatal deaths by up to 8 per 1000 births compared to 4 visits
- More contacts = more opportunities to detect and manage complications (preeclampsia, growth restriction, anaemia, infections)
- No significant difference in outcomes was found between 8 contacts and 11-15 contacts, so 8 was chosen as the practical minimum
What Happens at These Contacts?
The guidelines contain 49 recommendations covering:
- Blood tests (Hb, blood group, VDRL, HIV, urine dipstick)
- Ultrasound (ideally at <24 weeks)
- Nutritional counselling (iron 30-60 mg/day + folic acid 400 mcg/day)
- Tetanus toxoid vaccination
- BP monitoring for preeclampsia
- Fetal growth assessment
- Screening for malaria/HIV (where endemic)
- Mental health and domestic violence screening
Key Points to Remember
| Old Model | New Model |
|---|---|
| 4 visits (FANC, pre-2016) | 8 contacts (2016 onwards) |
| "Visit" terminology | "Contact" terminology |
| Minimum standard | Evidence-based optimum |
Source: WHO ANC Guidelines 2016
Mechanism of pre eclampsia
Finding Sources
Searching PubMed
preeclampsia pathogenesis sFlt-1 PlGF angiogenic
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Excellent - textbook diagrams retrieved. Here is the comprehensive mechanism of pre-eclampsia:
Mechanism of Pre-eclampsia
Pre-eclampsia is a two-stage disease originating in the placenta and culminating in widespread maternal endothelial dysfunction.
STAGE 1 - Defective Placentation (Weeks 8-18)
Normal Placentation
In a normal pregnancy, extravillous cytotrophoblasts invade the uterine spiral arteries of the decidua and myometrium. They replace the endothelial layer, transforming narrow resistance vessels into wide, flaccid, high-capacitance vessels - enabling the massive increase in uteroplacental blood flow needed to support the fetus.
What Goes Wrong in Pre-eclampsia
Cytotrophoblast invasion is shallow - limited only to the superficial decidua. The myometrial segments remain narrow and undilated. This occurs because:
- Cytotrophoblasts fail to undergo pseudovasculogenesis - the normal process of switching from an epithelial adhesion molecule phenotype to an endothelial one
- HIF-1 (Hypoxia Inducible Factor-1) is overactive, upregulating TGF-β3, which blocks trophoblast invasion
- HLA-G expression by cytotrophoblasts is reduced or absent, impairing immune tolerance at the maternal-fetal interface
- Decidual NK cells, which normally promote angiogenesis and trophoblast invasion, are dysfunctional
- Polymorphisms in KIR receptors (NK cells) and HLA-C (trophoblasts) may genetically predispose to this failure
Result: Inadequate spiral artery remodeling → placental ischemia and hypoperfusion → the ischemic/dysfunctional placenta releases toxic mediators into the maternal circulation.
STAGE 2 - Maternal Syndrome (Weeks 20+)
The Central Mechanism: Angiogenic Imbalance
The ischemic placenta releases excess anti-angiogenic factors:
| Factor | What it is | Effect |
|---|---|---|
| sFlt-1 (soluble fms-like tyrosine kinase-1) | Truncated splice variant of VEGFR-1 (Flt1) - lacks transmembrane domain | Scavenges free VEGF and PlGF in the circulation, preventing them from binding their endothelial receptors |
| sEng (soluble endoglin) | Truncated TGF-β receptor | Inhibits TGF-β1 and TGF-β3 signaling; potentiates sFlt-1's effects |
Pro-angiogenic factors are simultaneously depleted:
- Free VEGF (vascular endothelial growth factor) - falls
- Free PlGF (placental growth factor) - falls markedly (used clinically as a biomarker)
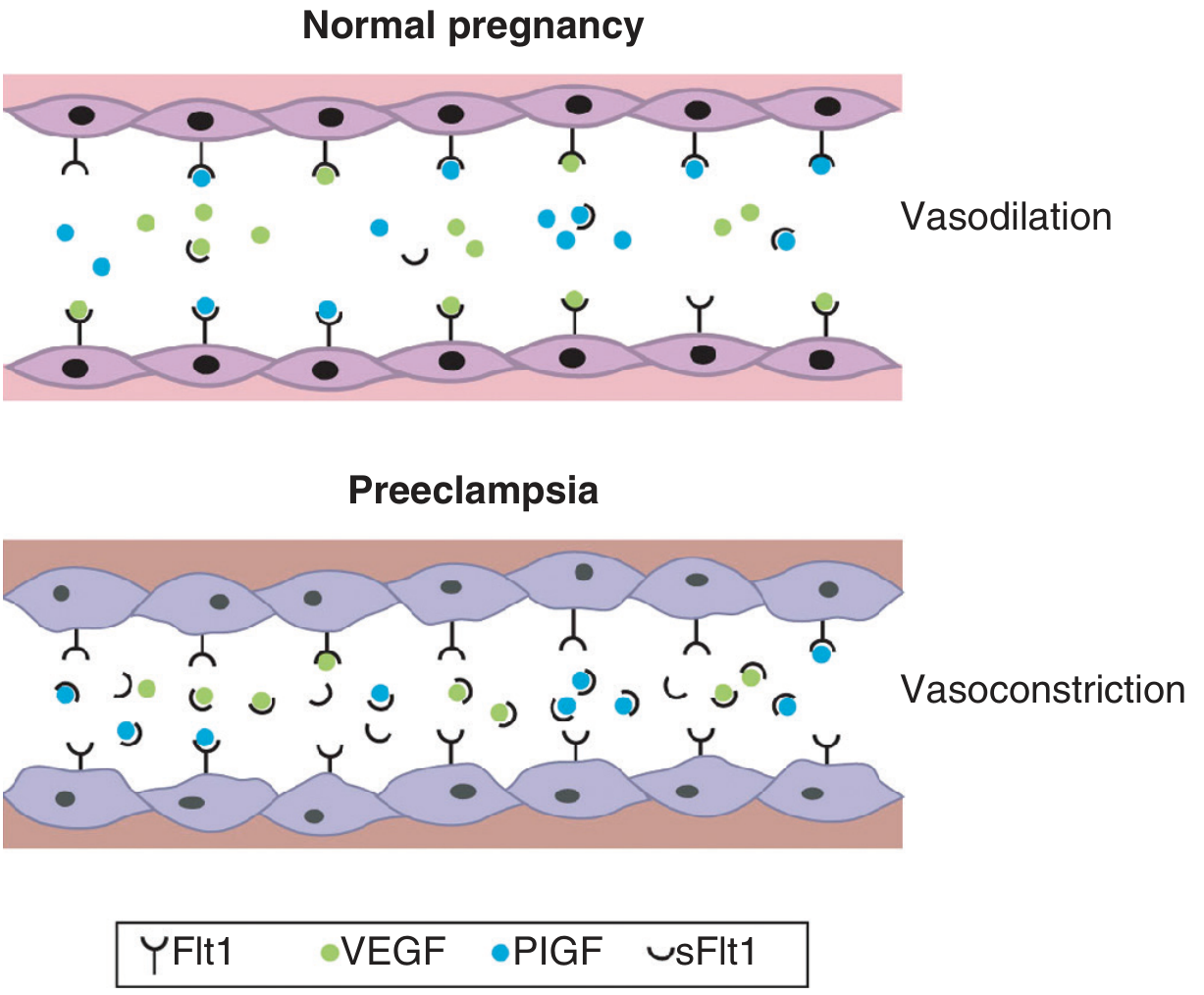
In normal pregnancy, VEGF and PlGF freely signal through Flt1 receptors, maintaining vasodilation. In pre-eclampsia, excess sFlt1 traps these molecules, causing vasoconstriction and endothelial dysfunction.
Maternal Endothelial Dysfunction
Without adequate VEGF/PlGF signaling, the maternal endothelium is damaged systemically:
- Impaired nitric oxide (NO) production → vasoconstriction
- Impaired prostacyclin (PGI₂) synthesis → platelet aggregation
- Increased endothelin, angiotensin II sensitivity → further vasoconstriction
- Activation of coagulation (fibrin deposition)
- Endothelial activation markers elevated: von Willebrand antigen, cellular fibronectin, soluble E-selectin, ICAM-1
Overall Pathogenesis Diagram
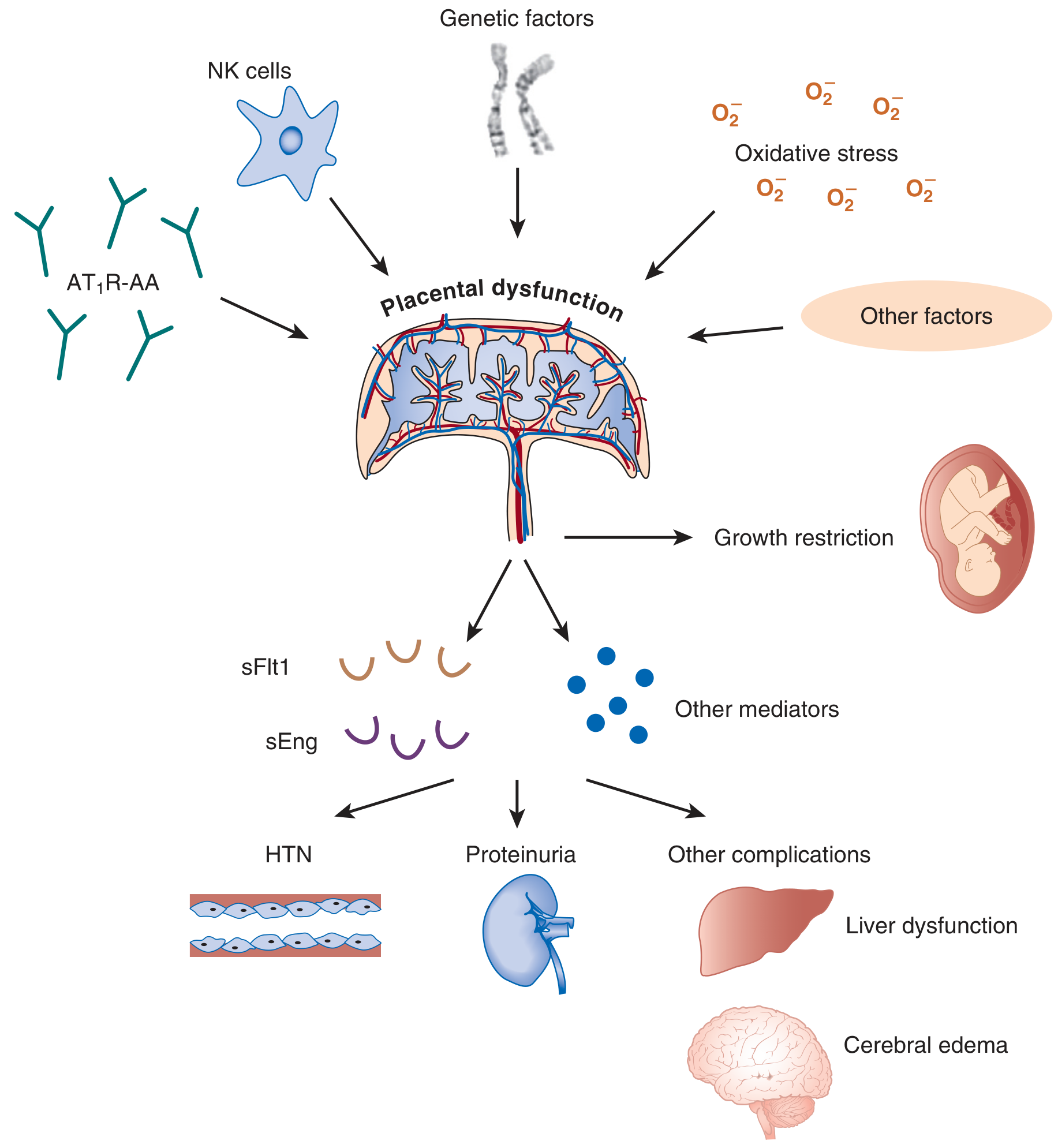
Genetic factors, NK cell dysfunction, angiotensin receptor autoantibodies (AT₁R-AA), and oxidative stress all converge on placental dysfunction → release of sFlt1 and sEng → hypertension, proteinuria, liver dysfunction, cerebral edema.
How Each Organ Is Affected
1. Kidney - Proteinuria (Glomerular Endotheliosis)
- VEGF is essential for maintaining the fenestrated endothelium of glomerular capillaries
- sFlt1 depletes VEGF → glomerular endothelial cells swell and vacuolate, capillary lumen is obliterated
- This lesion is called glomerular endotheliosis - pathognomonic of pre-eclampsia
- Result: proteinuria (≥300 mg/24 hours), reduced GFR
2. Vasculature - Hypertension
- Loss of VEGF-mediated NO production + endothelin excess → severe vasoconstriction
- High SVR, low cardiac output (opposite of normal pregnancy)
- Exaggerated sensitivity to angiotensin II and norepinephrine
3. Brain - Seizures / Eclampsia
- Endothelial dysfunction → breakdown of blood-brain barrier → vasogenic cerebral edema
- Predominantly in parieto-occipital white matter (Posterior Reversible Encephalopathy Syndrome - PRES)
- Note: cerebral edema correlates with endothelial damage markers, NOT with BP level
4. Liver - HELLP Syndrome
- Endothelial damage → fibrin deposition in hepatic sinusoids → periportal necrosis
- HELLP = Hemolysis, Elevated Liver enzymes, Low Platelets
- Microangiopathic hemolysis from fibrin strands shearing RBCs
Other Contributing Mechanisms
| Mechanism | Detail |
|---|---|
| Oxidative stress | Excess ROS from ischemic placenta damages maternal endothelium (likely secondary, not primary) |
| Immunologic intolerance | First pregnancy or new partner = novel paternal antigens → abnormal immune response → impaired trophoblast invasion |
| Angiotensin receptor autoantibodies (AT₁R-AA) | Activate AT₁ receptors on trophoblasts and vascular smooth muscle → vasoconstriction + placental dysfunction |
| H. pylori | Higher prevalence in hyperemesis gravidarum cases; may worsen gastric dysmotility |
| Genetic factors | Polymorphisms in STOX1, factor V Leiden, MTHFR, complement genes |
Summary: The Two-Stage Model
Stage 1 (Asymptomatic, Weeks 8-18):
Shallow trophoblast invasion
→ Undilated spiral arteries
→ Placental ischemia + hypoxia
Stage 2 (Symptomatic, Week 20+):
Ischemic placenta releases sFlt-1 + sEng
→ Depletion of free VEGF + PlGF
→ Widespread maternal endothelial dysfunction
→ HTN + proteinuria + HELLP + seizures
Sources: Brenner & Rector's The Kidney, 10th ed. (most detailed mechanistic source); Creasy & Resnik's Maternal-Fetal Medicine. Recent systematic review [PMID 38674114]: Chiang et al., 2024, confirms the sFlt-1/PlGF angiogenic imbalance as the dominant molecular mechanism.
Degrees of consanguineous marriage
Finding Sources
Searching the Web
degrees of consanguineous marriage coefficient of inbreeding first second cousin genetics
Finding Sources
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.