Toxicosis in pregnancy
Reading File
Finding Sources
Finding Sources
Searching PubMed
preeclampsia management guidelines
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Reading File
Reading File
I now have comprehensive information to write a thorough answer. Let me compile this.
Toxicosis in Pregnancy
The term "toxicosis of pregnancy" (also called gestosis or toxemia) encompasses two main clinical syndromes:
- Early toxicosis - nausea, vomiting, and hyperemesis gravidarum (first trimester)
- Late toxicosis - hypertensive disorders including preeclampsia, eclampsia, and HELLP syndrome (second/third trimester)
Part 1: Early Toxicosis - Nausea, Vomiting & Hyperemesis Gravidarum
Nausea and Vomiting of Pregnancy (NVP)
Nausea affects up to 80% of pregnancies; when severe it progresses to hyperemesis gravidarum.
Hyperemesis Gravidarum
Definition: Intractable nausea/vomiting causing:
- Weight loss >5% of total body weight
- Dehydration
- Starvation metabolism with prolonged ketonemia and ketonuria
Incidence: Approximately 1% of pregnancies
Etiology:
- Rising estradiol and hCG levels
- Maternal cytokines
- Helicobacter pylori infection (significantly associated; treating H. pylori decreases vomiting)
- Studies suggest early treatment of NVP may prevent progression to hyperemesis
Complications:
- Wernicke encephalopathy (vitamin B1 deficiency) - in the mother
- Bleeding diathesis (vitamin K deficiency) - in the fetus
- Abnormal liver function (bilirubin, alkaline phosphatase elevations that resolve with treatment)
Diagnostic workup:
- Urinalysis (ketones, specific gravity, infection)
- Serum chemistry (hypokalemia, contraction alkalosis, elevated anion gap)
- Liver function tests
Management:
- Rehydration: 2 L Ringer's lactate IV at 500 mL/hr
- Thiamine (Vitamin B1) BEFORE any dextrose - to prevent Wernicke encephalopathy
- Then dextrose-containing IV fluids (D5/0.45NS) until ketones clear
- Antiemetics: Diclegis (doxylamine + B6) is first line
- Potassium and magnesium repletion as needed
- Refractory cases: methylprednisolone 16 mg PO/IV every 8 hours for 3 days (last resort)
- If weight cannot be maintained: nasogastric enteral nutrition
- Rosen's Emergency Medicine, p. 3364
Part 2: Late Toxicosis - Hypertensive Disorders of Pregnancy
Overview
Hypertensive disorders of pregnancy (gestational hypertension, preeclampsia, eclampsia) affect approximately 85 per 1000 deliveries in the United States. Preeclampsia occurs in 3-5% of pregnancies overall.
Preeclampsia
Pathogenesis
The central problem is failed remodeling of uterine spiral arteries. In normal pregnancy, trophoblasts invade and remodel the spiral arteries into wide, low-resistance sinusoids. In preeclampsia, this remodeling fails - the arteries remain narrow and muscular, causing placental hypoxia.
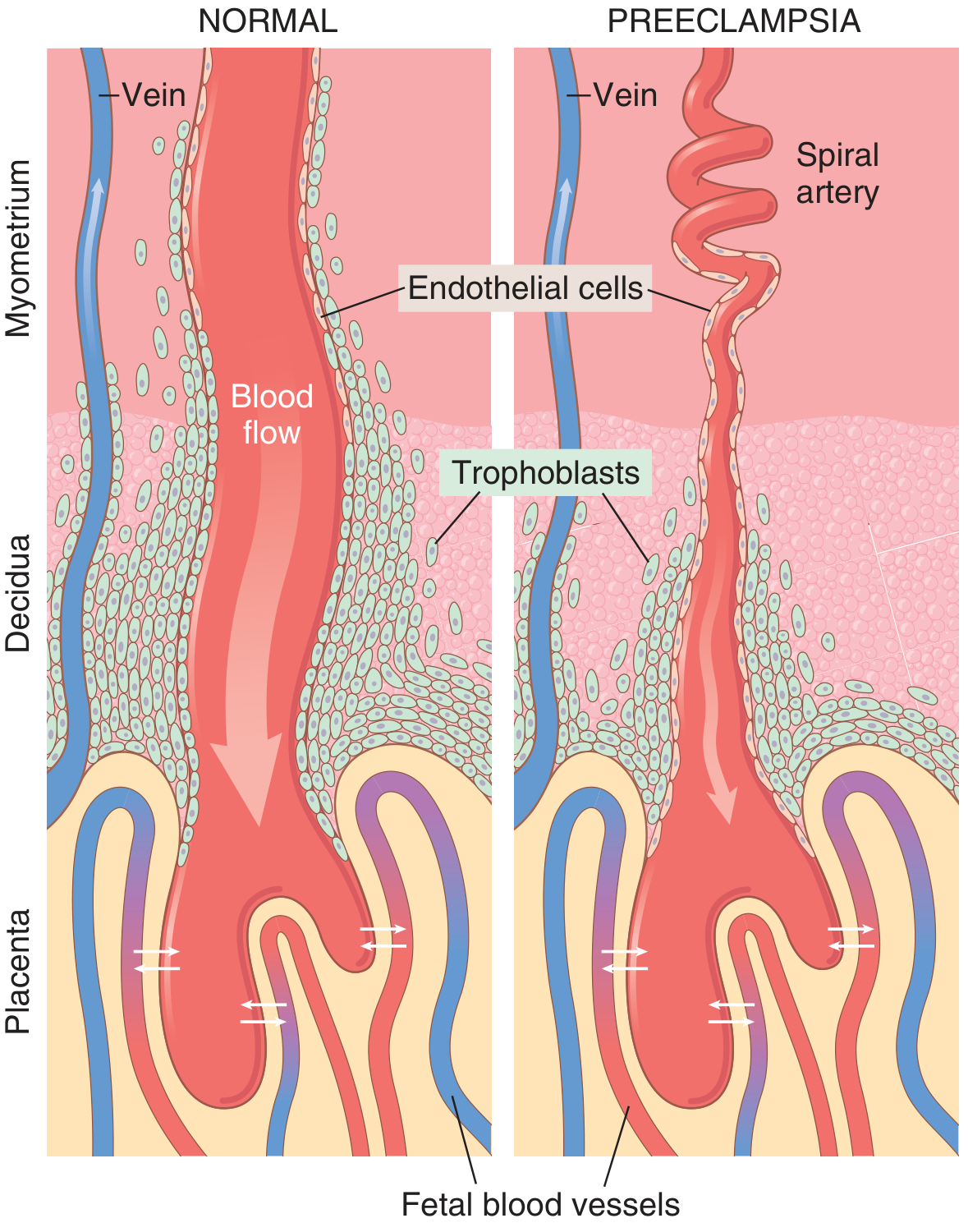
In normal pregnancy (left), trophoblasts fully invade and widen the spiral arteries. In preeclampsia (right), the arteries remain narrow and tortuous, restricting blood flow to the placenta.
The ischemic placenta then releases factors into the maternal circulation:
- Anti-angiogenic proteins: soluble FMS-like tyrosine kinase-1 (sFlt-1) and soluble endoglin
- These antagonize VEGF and TGF-β, causing systemic maternal endothelial dysfunction
- Inflammatory cytokines (TNF-α, IL-6) amplify the response
Consequences of endothelial dysfunction:
| Mechanism | Effect |
|---|---|
| ↓ prostacyclin (PGI2) + ↑ thromboxane A2 | Hypertension |
| ↓ endothelial antithrombotic factors | Hypercoagulability |
| Glomerular endotheliosis | Proteinuria, reduced GFR |
| Vascular barotrauma in brain | Cerebral edema, PRES, seizures |
| Hepatic arteriolar spasm + hemorrhage | Liver failure, HELLP |
- Robbins & Kumar Basic Pathology, p. 702
- Guyton & Hall Medical Physiology, p. 1041
Diagnostic Criteria
Preeclampsia is diagnosed at ≥20 weeks gestation with:
- Blood pressure ≥ 140/90 mmHg on two occasions 4 hours apart
- Plus proteinuria OR evidence of end-organ damage (thrombocytopenia, renal insufficiency, impaired liver function, pulmonary edema, new-onset headache/visual disturbances)
Gestational hypertension = BP ≥140/90 without proteinuria or end-organ damage. A temporary diagnosis; if preeclampsia doesn't develop, the label is retained.
Risk Factors
- Nulliparity (greatest population attributable fraction: 32.3%)
- Prior preeclampsia, antiphospholipid syndrome, chronic hypertension (25% develop preeclampsia), chronic renal disease
- Pregestational diabetes mellitus (20% overall; up to 70% in White's classes F/R)
- Obesity (increases risk ~3-fold; accounts for up to 1/3 of US cases)
- Multiple gestation (6.7% twins, 12.7% triplets, 20% quadruplets)
- Hydatidiform mole (70%!), extremes of maternal age, Black race (for severe forms)
- Creasy & Resnik's Maternal-Fetal Medicine
Signs and Symptoms of Severe Preeclampsia
- Cerebral: headache, dizziness, tinnitus, drowsiness, altered mental status
- Visual: blurred vision, scotomata, diplopia, amaurosis
- GI: nausea, vomiting, epigastric/RUQ pain (hepatic involvement)
- Renal: oliguria, anuria, hematuria
Organ Pathology
Brain: Cerebral edema on CT/MRI; Posterior Reversible Encephalopathy Syndrome (PRES) - parietooccipital vasogenic edema, typically reversible. Long-term: increased white matter lesions, cognitive dysfunction even after recovery.
Liver: Visible gross lesions in 60% of eclamptic women. Two phases:
- Hemorrhage into hepatic cellular columns (vasodilation)
- Hepatic infarction (vasospasm)
Kidneys: Glomerular endotheliosis - pathognomonic finding. Glomeruli show decreased capillary lumen diameter, increased endothelial-mesangial cell volume, reduced GFR.
Placenta: Infarcts (more numerous than in normal pregnancy), retroplacental hemorrhage, ischemic villi with syncytial knots, fibrinoid necrosis of decidual vessels (acute atherosis).
HELLP Syndrome
Develops in 5-10% of patients with preeclamptic symptoms and represents a life-threatening complication:
| Letter | Stands for | Laboratory finding |
|---|---|---|
| H | Hemolysis | Microangiopathic hemolytic anemia on peripheral smear |
| EL | Elevated Liver enzymes | ↑ AST, ALT, LDH |
| LP | Low Platelets | Thrombocytopenia (platelet consumption) |
May progress to DIC. Treatment: prompt delivery. Early recognition is critical - women presenting in the third trimester with hypertension, nausea/vomiting, or RUQ pain must have CBC with platelets and LFTs before discharge.
Eclampsia
Eclampsia = preeclampsia + seizures or coma
- Occurs in approximately 0.2% of pregnancies
- Mechanism: hypertensive encephalopathy, loss of cerebral vascular autoregulation
- Perinatal mortality: 2-8.6%
- Maternal mortality: <2%, mainly from intracranial hemorrhage
Management
Antihypertensive Therapy
National US guidelines mandate aggressive treatment of:
- Systolic BP ≥ 160 mmHg, OR
- Diastolic BP ≥ 110 mmHg, if persistent for ≥15 minutes
Agents used (progressive IV protocol):
- IV labetalol
- IV hydralazine
- Oral nifedipine
Undertreated hypertension (especially with low/borderline platelets) is the greatest risk factor for intracranial hemorrhage.
Seizure Prevention/Treatment
Magnesium sulfate (MgSO₄) is the treatment of choice for seizure prophylaxis and eclampsia:
- Loading dose: 4-6 g IV over 15 minutes
- Maintenance: 1-3 g/hr continuous infusion
- Monitor: deep tendon reflexes, urine output, serum Mg levels
- Antidote: IV calcium gluconate
Definitive Treatment
Delivery is the only cure. In preterm pregnancies, risks of prematurity are weighed against severity of preeclampsia. Postpartum preeclampsia can present in the emergency department and requires the same BP management.
Special Situations
-
HELLP syndrome and severe preeclampsia with multisystem dysfunction: maternal-fetal medicine subspecialist consultation
-
Inadequate local facilities: transfer to tertiary care center
-
Creasy & Resnik's Maternal-Fetal Medicine, pp. 1057-1060
-
Textbook of Family Medicine, p. 497
Prevention
Low-dose aspirin (initiated in the first trimester) is recommended for women at high risk of preeclampsia. Calcium supplementation may reduce risk in calcium-deficient populations.
Summary Classification
| Disorder | Onset | Key Features | Treatment |
|---|---|---|---|
| Hyperemesis gravidarum | 1st trimester | Vomiting, >5% weight loss, ketonuria | IV fluids, thiamine, antiemetics |
| Gestational hypertension | ≥20 weeks | BP ≥140/90, no proteinuria/end-organ damage | Monitoring, antihypertensives if severe |
| Preeclampsia | ≥20 weeks (usually 3rd trimester) | Hypertension + proteinuria/end-organ damage | Antihypertensives, MgSO₄, delivery |
| Severe preeclampsia | Same | BP ≥160/110, multi-organ involvement | Urgent delivery, ICU-level care |
| HELLP syndrome | 3rd trimester/postpartum | Hemolysis + ↑LFTs + ↓platelets | Emergency delivery |
| Eclampsia | 3rd trimester/postpartum | Seizures/coma + preeclampsia criteria | MgSO₄, deliver |
Explain the pathogenesis of early toxicosis in pregnancy as a pathologist understands it
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
I've exhausted the database queries but have gathered comprehensive pathology content. Now I can write the detailed pathologist-oriented answer.
Pathogenesis of Early Toxicosis in Pregnancy - A Pathologist's Perspective
Early toxicosis refers to the nausea and vomiting of pregnancy (NVP) and its severe form, hyperemesis gravidarum (HG), occurring predominantly in weeks 4-16 of gestation. From a pathologist's standpoint, this is not a single disease but the clinical output of multiple converging biological mechanisms - hormonal, neural, immunological, infectious, and genetic - all centering on the profound physiological upheaval of early pregnancy.
I. Epidemiological Framework (the Starting Point for Pathological Thinking)
- NVP affects 60-70% of all pregnant women; vomiting in >40%
- Onset: weeks 4-6; peak: weeks 8-12; resolution: week 20 in most
- Hyperemesis gravidarum (the severe, pathological end): <2% of pregnancies, defined by ketonuria + >5% weight loss from pre-pregnancy weight
- Recurs in subsequent pregnancies in 15-19% of previously affected women
The timing is not random - it precisely mirrors the period of maximum placental trophoblast proliferation and the surge of pregnancy-specific hormones, giving a strong mechanistic clue.
- Sleisenger & Fordtran's Gastrointestinal and Liver Disease, p. 655
- Creasy & Resnik's Maternal-Fetal Medicine, p. 1564
II. The Central Pathological Mechanisms
1. The hCG Hypothesis (Primary Driver)
Human chorionic gonadotropin (hCG) is the strongest candidate for the primary trigger of early toxicosis. The evidence from a pathologist's view is compelling:
Supporting evidence (correlation = near-causation here):
- Symptoms worsen precisely during the period of peak hCG concentrations (weeks 8-12)
- Conditions with higher hCG levels are universally associated with worse NVP and higher rates of HG:
- Multiple gestation
- Gestational trophoblastic disease (hydatidiform mole)
- Trisomy 21 (which produces excess hCG)
- Hydrops fetalis
- Symptoms improve as hCG falls in the second trimester
Mechanism: The alpha subunit of hCG shares structural homology with TSH (thyroid-stimulating hormone). This cross-reactivity causes:
- Transient gestational thyrotoxicosis - hCG stimulates the TSH receptor, suppresses endogenous TSH, and elevates free T4
- Abnormal thyroid function tests are found in two-thirds of HG patients
- The resulting mild hyperthyroid state amplifies nausea (thyroid hormones modulate gastric motility and the central vomiting pathways)
- Importantly, this is not true hyperthyroidism - it resolves with delivery and does not require antithyroid treatment
2. Estrogen and Progesterone Effects on GI Motility
Estrogen and progesterone act directly on smooth muscle and the enteric nervous system:
| Hormone | GI Effect | Pathological Consequence |
|---|---|---|
| Progesterone | Relaxes smooth muscle, decreases LES tone | Delayed gastric emptying, gastric stasis, reflux |
| Estrogen (rising) | Slows GI transit time, alters gastric motility | Prolonged exposure of gut to food, heightened sensitivity |
| Both | Decrease lower esophageal sphincter pressure | GERD, regurgitation, nausea amplification |
Elevated estrogen concentrations (as seen in obesity) are independently associated with more severe HG - obese women have higher baseline estrogen and markedly worse symptoms.
The progesterone-mediated decrease in resting lower esophageal sphincter pressure is the dominant mechanism behind pregnancy-associated GERD, which compounds nausea through esophageal irritation and vagal activation.
- Sleisenger & Fordtran's GI & Liver Disease, p. 655
3. The Gut-Derived Hormones: Ghrelin and Leptin
These satiety and appetite-regulating hormones from the gastrointestinal tract and adipose tissue are disrupted in pregnancy:
- Ghrelin (the "hunger hormone," produced in the gastric fundus): levels are altered in HG; its role in gastric motility means changes directly affect the rate of gastric emptying
- Leptin (produced by adipose tissue and the placenta): placental leptin secretion rises in early pregnancy and has been implicated in modulating the nausea response through central hypothalamic pathways
- Both are currently considered contributing factors rather than primary causes
4. The Central Vomiting Pathways - Neural Architecture of HG
From a neuropathological standpoint, the vomiting arc involves two central structures:
A. The Vomiting Center (nucleus tractus solitarius / NTS complex):
- Located in the medulla oblongata
- Receives afferent signals from the vagus nerve (gut), vestibular apparatus, higher cortical centers, and the area postrema
- Coordinates the efferent motor act of vomiting via the phrenic nerve, spinal motor neurons, and cranial nerves
B. The Area Postrema (Chemoreceptor Trigger Zone, CTZ):
- Located at the floor of the fourth ventricle
- Lies outside the blood-brain barrier - directly sensitive to circulating chemical triggers (hCG, estrogen metabolites, toxins)
- Rich in dopamine (D2), serotonin (5-HT3), and substance P/NK1 receptors
- Circulating hCG and estrogen metabolites stimulate the CTZ → relay to the vomiting center → nausea and emesis
- This is why the nuclei of the solitary tract, raphe, and area postrema are thought to be the sites of steroid action (the mechanism by which corticosteroids rescue refractory HG)
Sensory hypersensitization:
- In HG, vomiting is triggered not only by chemical stimuli but by olfactory, auditory, and visual stimuli - reflecting a pathological lowering of the sensory threshold at the level of the NTS and cortical-limbic pathways
- Hyperolfaction (heightened smell sensitivity) is a hallmark symptom, consistent with increased CNS arousal of the emetic network
5. Helicobacter pylori - The Infectious Contribution
This is perhaps the most "classical pathology" element of early toxicosis:
- Two meta-analyses show significantly increased rates of H. pylori infection in pregnant women with HG vs. uncomplicated NVP
- Proposed mechanism: H. pylori colonizes the gastric antrum, induces local mucosal inflammation (neutrophil infiltration, IL-8, TNF-α release), alters gastric emptying, and directly stimulates the vagus nerve
- The inflammatory cytokines produced by H. pylori gastritis may amplify the central vomiting response through the area postrema
- Symptomatic improvement has been documented after H. pylori eradication during pregnancy using non-teratogenic regimens
- Importantly, H. pylori itself does not cause HG, but acts as a disease modifier - amplifying the hormonal and neural triggers already in place
6. Psychoneuroimmunological and Psychosocial Factors
From a pathobiological perspective, the psychological component is not "psychosomatic" in the dismissive sense:
- Familial clustering of HG strongly implies a genetic predisposition - likely in genes governing hCG receptor sensitivity, serotonin transporter function, or central emetic threshold
- Psychological stress activates the hypothalamic-pituitary-adrenal (HPA) axis and alters GI motility via corticotropin-releasing hormone (CRH) - which also acts on peripheral mast cells in the gut mucosa, increasing permeability and enteric nerve excitability
- Women with prior psychiatric disorders have higher rates of HG hospitalization - though causality is bidirectional (HG causes psychological distress)
III. Pathological Consequences of Severe HG (What the Pathologist Sees)
When early toxicosis is severe and untreated, the systemic pathological effects are:
Metabolic/Biochemical
- Hypokalemia, hyponatremia - from vomiting losses
- Contraction metabolic alkalosis - from loss of HCl
- Ketonemia/ketonuria - starvation metabolism; fat catabolism
- Elevated anion gap - in prolonged starvation
- Hypoamylasemia correction note: Hyperamylasemia occurs in ~25% of HG cases - caused by excessive salivary gland production stimulated by prolonged vomiting (not pancreatitis)
- Transaminase elevation (AST, ALT) in 25-40% + mild hyperbilirubinemia - from hepatic dysfunction secondary to malnutrition and fatty change; fully reversible
Neurological (Wernicke Encephalopathy)
- Thiamine (B1) is water-soluble and depleted rapidly when oral intake ceases
- Thiamine is an essential cofactor for pyruvate dehydrogenase, alpha-ketoglutarate dehydrogenase, and transketolase - all required for aerobic glucose metabolism
- Administration of dextrose (glucose) WITHOUT thiamine first drives pyruvate through glycolysis, depleting the remaining thiamine and precipitating Wernicke encephalopathy
- Pathology of Wernicke: hemorrhagic necrosis and petechiae in the mammillary bodies, periventricular gray matter, periaqueductal region, and floor of the 4th ventricle
- Classic triad: ophthalmoplegia, ataxia, confusion
Fetal Consequences
-
Vitamin K deficiency (fat-soluble, crosses placenta poorly when mother is deficient) → neonatal bleeding diatheses
-
Low birth weight, SGA, prematurity, low 5-minute Apgar scores in infants of HG women with <7 kg gestational weight gain
-
Rarely: esophageal Mallory-Weiss tears, pneumomediastinum (barotrauma from forceful vomiting), esophageal rupture (Boerhaave syndrome)
-
Sleisenger & Fordtran's GI & Liver Disease, p. 655
-
Creasy & Resnik's Maternal-Fetal Medicine, p. 1564
-
Rosen's Emergency Medicine, p. 3364
IV. Integrated Pathogenetic Model
Trophoblast proliferation
↓
↑↑ hCG secretion
↙ ↘
Thyroid Area Postrema
stimulation (CTZ)
(↑ free T4, stimulation
↓ TSH) ↓
↘ Vomiting Center
→ NTS/raphe ← (nucleus tractus
↑ solitarius)
↑ Estrogen ↑
↑ Progesterone Vagal input from
(↓ GI motility, H. pylori gastritis /
↓ LES tone) GI dysmotility
↓
NAUSEA / VOMITING
↓ (if severe / untreated)
Starvation → Ketonuria → Wernicke
Hypokalemia → Alkalosis → Fetal growth restriction
V. Why This Matters Pathologically
The key insight a pathologist brings is that early toxicosis is a hormonally driven, self-limited disease of trophoblast excess, with the severity determined by:
- The degree of hCG/estrogen rise (amplified by multiple gestation, mole, trisomy 21)
- Individual receptor sensitivity (genetic variation in hCG receptor, serotonin transporter)
- Comorbid H. pylori gastritis (amplifies the afferent vagal signal)
- Central emetic threshold (modulated by genetics, prior experience, psychosocial state)
The fact that symptoms resolve with termination of pregnancy - or spontaneously as the placenta matures and hCG falls - is the clearest evidence of the hormonal-trophoblastic etiology. It is a disease of the placenta communicating with the maternal brain, mediated by hormones that cross into the circulation and directly activate the area postrema outside the blood-brain barrier.
This is a shared conversation. Sign in to Orris to start your own chat.