If insulin inhibits lipolysis, why does insulin resistance in type 2 diabetes increase lipolysis and free fatty acids, leading to fatty liver despite high insulin levels?”
Reading File
Finding Sources
Finding Sources
Searching PubMed
insulin resistance adipose lipolysis fatty liver selective resistance
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
I now have all the information needed for a thorough, well-sourced answer. Let me compile it.
Why Insulin Resistance Increases Lipolysis and Causes Fatty Liver Despite High Insulin Levels
This is one of the most instructive paradoxes in metabolic medicine. The resolution lies in three interlocking concepts: (1) the extreme sensitivity of adipose anti-lipolysis to insulin resistance, (2) selective/partial insulin resistance in the liver, and (3) the amplifying feedback loop between free fatty acids (FFAs) and insulin resistance itself.
1. Normal Insulin Action on Fat - How it Works
In healthy physiology, insulin suppresses lipolysis in adipose tissue through two complementary mechanisms:
- Inhibiting hormone-sensitive lipase (HSL) and adipose triglyceride lipase (ATGL): Insulin activates phosphodiesterase, which breaks down cAMP. Without cAMP, protein kinase A (PKA) is not activated, so HSL is not phosphorylated and remains inactive. ATGL (which initiates the first step of triglyceride hydrolysis) is similarly suppressed.
- Activating lipoprotein lipase (LPL) in adipose tissue: This promotes uptake of circulating triglycerides into fat stores rather than their release.
The net result: FFAs stay inside adipocytes, plasma FFA levels fall after meals. (Basic Medical Biochemistry, p. 1272; Ganong's Review of Medical Physiology)
2. The Key Paradox - Anti-Lipolysis is the Most Sensitive Pathway to Insulin Resistance
As Harrison's states explicitly:
"The inhibition of lipolysis in adipose tissue is the most sensitive pathway of insulin action. Thus, when insulin resistance develops, increased lipolysis produces more fatty acids, which further decreases the anti-lipolytic effect of insulin."
- Harrison's Principles of Internal Medicine 22E, Insulin Resistance section
This means that anti-lipolysis fails first and most completely when insulin resistance develops. Even low concentrations of insulin would normally suppress lipolysis - but in insulin resistance, even the massively elevated compensatory insulin cannot fully suppress it. So adipocytes release FFAs in an unregulated, fasting-like manner, all day long - even in the fed (postprandial) state.
The molecular mechanism: insulin resistance impairs the PI3K/Akt signaling cascade downstream of the insulin receptor. Akt normally phosphorylates and activates phosphodiesterase 3B (PDE3B), which degrades cAMP and keeps PKA (and thus HSL) inactive. With impaired Akt signaling, PDE3B stays underactivated, cAMP stays elevated, HSL remains active, and lipolysis runs unchecked.
3. The Self-Amplifying Feedback Loop
FFAs are not just a product of lipolysis - they cause and worsen insulin resistance, creating a vicious cycle:
- Excess FFA flux into muscle impairs IRS-1/PI3K/Akt insulin signaling by activating protein kinase C (PKC-theta), DAG, and ceramide pathways
- This further reduces glucose uptake in muscle
- The pancreas compensates by secreting even more insulin (compensatory hyperinsulinemia)
- More glucose remains in circulation, driving more de novo lipogenesis in the liver
- TNF-alpha and IL-6 from adipose macrophages (in expanded visceral fat) further stimulate lipolysis and worsen insulin signaling
As Lippincott Biochemistry states: "Increased adipose lipolysis with production of free fatty acids is a hallmark of insulin resistance, and these FFAs are central mediators of cellular insulin resistance." (Biochemistry 8th ed. Lippincott, p. 949)
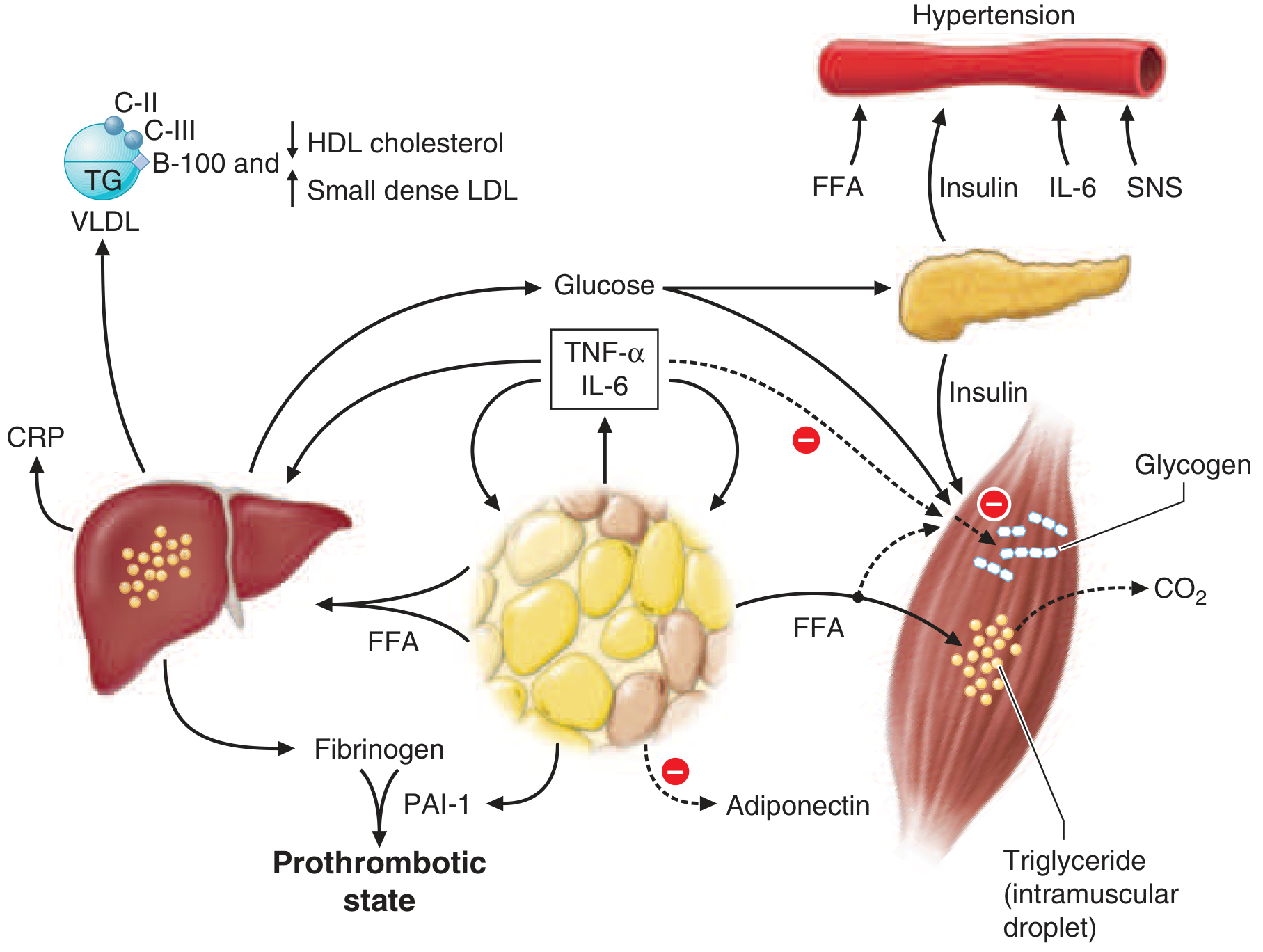
Figure from Harrison's 22E: FFAs flood from insulin-resistant adipose tissue into liver and muscle, producing the full metabolic syndrome.
4. Why the Liver Gets Fat - Selective Hepatic Insulin Resistance
This is the conceptual core of the paradox. The liver experiences selective (partial) insulin resistance:
| Hepatic Pathway | Insulin Sensitivity in T2D | Result |
|---|---|---|
| Suppression of gluconeogenesis | RESISTANT | Fasting hyperglycemia |
| Activation of SREBP-1c / de novo lipogenesis (DNL) | RETAINED (relatively) | Increased fat synthesis |
| VLDL secretion | Partially retained | Hypertriglyceridemia |
Harrison's explains: "Insulin resistance in the liver reflects the failure of hyperinsulinemia to suppress gluconeogenesis... As a result of insulin resistance in adipose tissue, lipolysis and free fatty acid flux from adipocytes are increased and efficiently cleared by the liver, leading to increased VLDL-triglyceride synthesis in hepatocytes." (Harrison's 22E, p. 722)
The liver becomes the central recipient of the FFA flood from insulin-resistant adipose tissue. It handles the excess by:
- Esterifying FFAs into triglycerides - this is the steatosis (fat droplets visible on biopsy/ultrasound)
- Packaging triglycerides into VLDL for export - causes hypertriglyceridemia
- Activating de novo lipogenesis (DNL) via SREBP-1c and ChREBP - the hyperinsulinemia that fails to suppress gluconeogenesis continues to activate SREBP-1c (sterol regulatory element-binding protein-1c) through a different PI3K branch, and excess glucose activates ChREBP (carbohydrate response element-binding protein). Together they upregulate fatty acid synthase (FAS), ACC1, and DGAT1/2.
Yamada's gastroenterology textbook quantifies this: in normal physiology, DNL contributes only 5-8% of hepatic triglyceride fatty acids in the fasted state. In NAFLD/insulin resistance, this rises to over 20%. (Yamada's Textbook of Gastroenterology 7e, p. 2062)
When triglyceride formation exceeds VLDL export capacity, fat accumulates - giving you metabolic-associated steatotic liver disease (MASLD, formerly NAFLD).
5. Three Sources Converge on the Liver Simultaneously
| Source | Mechanism | Contribution to Hepatic Fat |
|---|---|---|
| Adipose FFA release | Insulin-resistant HSL/ATGL runs unchecked | ~60% of hepatic fat in NAFLD |
| Diet | Chylomicron remnant uptake | ~15-20% |
| Hepatic DNL | SREBP-1c + ChREBP activated by insulin + glucose | ~20-26% (vs. 5-8% normally) |
6. Why High Insulin Doesn't Fix It
The critical reason the body cannot self-correct:
- The branch of insulin signaling that suppresses gluconeogenesis (via FOXO1) and activates anti-lipolysis (via PDE3B) becomes selectively resistant (likely through IRS-1 serine phosphorylation, a stress-mediated mechanism driven by FFAs, DAG, and ceramide)
- The branch of insulin signaling driving lipogenesis (via SREBP-1c through mTORC1) remains paradoxically intact or even upregulated
- The result: high insulin levels cannot suppress glucose production or lipolysis, but they do keep pushing fat synthesis - the worst of both worlds
Summary - The Paradox Resolved
Obesity/excess calories
↓
Expanded visceral adipose (↑ TNF-α, IL-6, ↓ adiponectin)
↓
IRS-1 serine phosphorylation → impaired PI3K/Akt signaling
↓
Anti-lipolysis FAILS (most sensitive pathway)
↓
Unrestrained HSL/ATGL → FFA flood into portal circulation
↓ ↓
FFA further worsens Liver: SREBP-1c/ChREBP still
insulin resistance activated by hyperinsulinemia
(muscle, liver, adipose) ↓
↑ DNL + FFA esterification
Pancreas ↑↑ insulin ↓
(compensatory) Triglyceride accumulation
↓
FATTY LIVER (MASLD)
Key sources:
- Harrison's Principles of Internal Medicine 22E - Insulin Resistance, Ch. 415 & 420
- Yamada's Textbook of Gastroenterology 7e - NAFLD pathogenesis, p. 2061-2062
- Lippincott Illustrated Reviews: Biochemistry 8e - Insulin resistance, p. 949-951
- Basic Medical Biochemistry 6e - Lipolysis regulation, p. 1272
In beta oxidation of fatty acids acyl co A dehydrogenase step is bypassed what does it mean
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
I have everything needed. Here is the complete, well-sourced answer:
"Bypassing" the Acyl-CoA Dehydrogenase Step in Beta-Oxidation
First - The Normal Beta-Oxidation Cycle (4 Steps)
Each round of beta-oxidation has exactly four reactions:
| Step | Enzyme | Reaction | Cofactor |
|---|---|---|---|
| 1 | Acyl-CoA dehydrogenase | Removes 2 H from C2-C3, creating a trans double bond | FAD → FADH₂ |
| 2 | Enoyl-CoA hydratase | Adds H₂O across the double bond | - |
| 3 | 3-Hydroxyacyl-CoA dehydrogenase | Oxidizes the -OH to a ketone | NAD⁺ → NADH |
| 4 | Thiolase | Cleaves with CoA-SH, releasing acetyl-CoA | CoA |
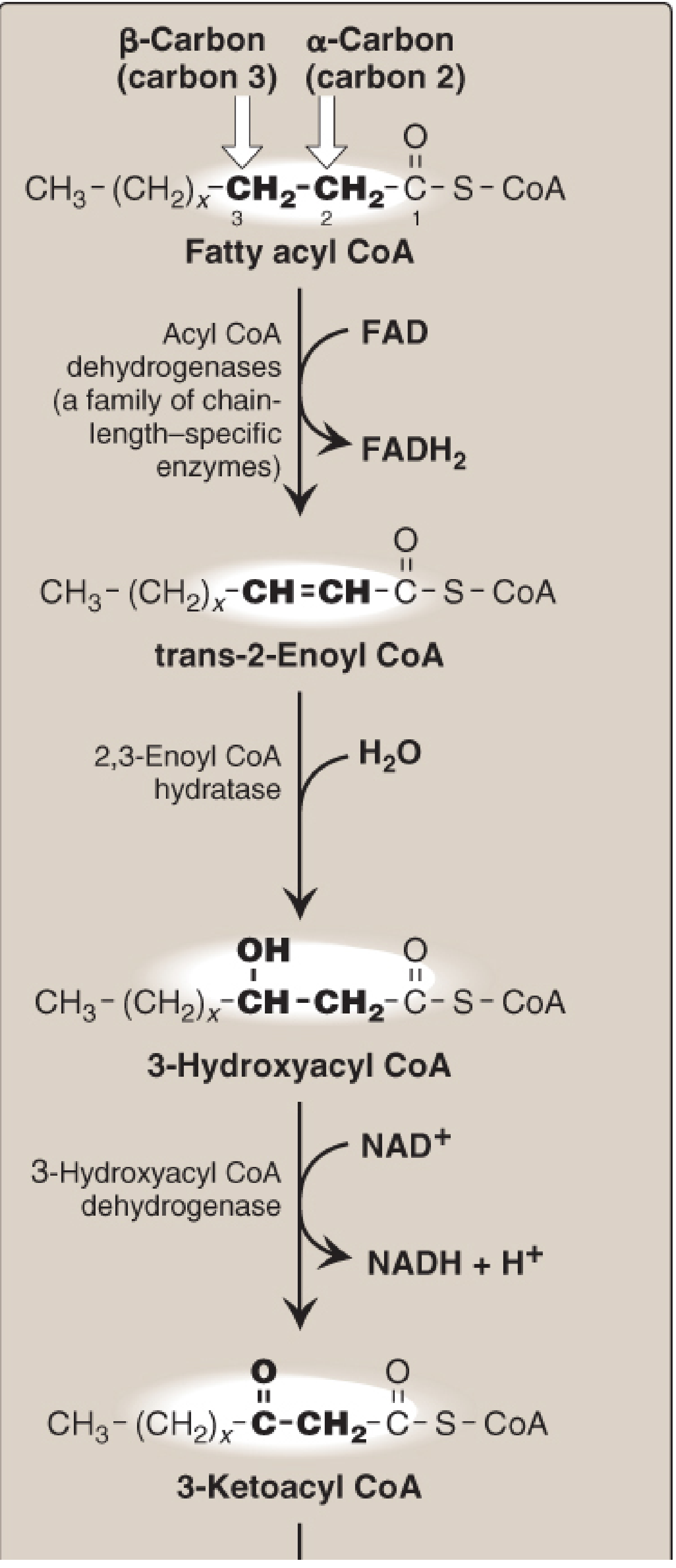
From Lippincott Illustrated Reviews: Biochemistry 8e - the four enzymes and intermediates of one beta-oxidation cycle
The first step (acyl-CoA dehydrogenase) has one job: introduce a trans-double bond between C2 (alpha) and C3 (beta). The product - trans-Δ2-enoyl-CoA - is what step 2 (enoyl-CoA hydratase) needs to act on.
What "Bypassing" Means
When a fatty acid already has a double bond between C2 and C3 in the correct trans configuration before that round of oxidation begins, the acyl-CoA dehydrogenase step is simply skipped - because the product it would make already exists. The chain enters the cycle at step 2 directly.
This happens when you are oxidizing unsaturated fatty acids (e.g., oleic acid 18:1, linoleic acid 18:2). These have pre-existing double bonds in their carbon chain. After one or more rounds of normal beta-oxidation, the chain gets shortened until that natural double bond ends up between C2 and C3 - at which point step 1 is redundant.
The Consequences of Bypassing - Why It Matters
1. One FADH₂ is LOST per bypass
The acyl-CoA dehydrogenase reaction produces FADH₂, which feeds electrons into the electron transport chain at Complex II (coenzyme Q), generating approximately 1.5 ATP.
When the step is bypassed, that FADH₂ is never made. So:
"Because unsaturated fatty acids are less reduced than saturated fatty acids, fewer reducing equivalents are produced by their oxidation."
- Lippincott Illustrated Reviews: Biochemistry 8e, p. 553
Net result: Each bypass costs you ~1.5 ATP compared to oxidizing a fully saturated fatty acid of the same length.
2. The pre-existing double bond must be in the RIGHT geometry
The enoyl-CoA hydratase (step 2) is specific - it only acts on a trans (2,3) double bond. Most natural unsaturated fatty acids have cis double bonds. So simply arriving at C2-C3 is not enough - the geometry must be corrected too.
This requires extra enzymes:
- Enoyl-CoA isomerase (for monounsaturated fatty acids like oleic acid 18:1Δ9): converts a cis-Δ3-enoyl-CoA to trans-Δ2-enoyl-CoA
- 2,4-Dienoyl-CoA reductase (NADPH-dependent) + isomerase (for polyunsaturated fatty acids like linoleic acid 18:2Δ9,12)
These enzymes essentially "fix" the geometry of the bypass product so it can be recognized by step 2. But note: the reductase uses NADPH, adding another small energetic cost.
Summary of the Bypass in One Diagram
Normal (saturated) fatty acid:
Acyl-CoA
↓ [Step 1: Acyl-CoA dehydrogenase + FAD → FADH₂] ← THIS STEP OCCURS
trans-Δ2-Enoyl-CoA
↓ [Step 2: Hydratase]
3-Hydroxyacyl-CoA
↓ [Step 3: Dehydrogenase → NADH]
3-Ketoacyl-CoA
↓ [Step 4: Thiolase → Acetyl-CoA]
Unsaturated fatty acid (after rounds of normal oxidation, double bond lands at C2-C3):
Acyl-CoA (already has trans double bond at C2-C3)
↓ [Step 1: BYPASSED - no enzyme needed, no FADH₂ produced]
trans-Δ2-Enoyl-CoA ← enters here directly
↓ [Step 2, 3, 4 proceed normally]
Clinical Relevance - ATP Yield Comparison
For palmitic acid (16:0, saturated):
- 7 complete cycles, 7 FADH₂ + 7 NADH + 8 acetyl-CoA → ~106 ATP net
For palmitoleic acid (16:1Δ9, one double bond):
- Same chain length, but 1 bypass → 1 fewer FADH₂ → loses ~1.5 ATP → ~104.5 ATP net
For linoleic acid (18:2) with two double bonds, the 2,4-dienoyl reductase step also consumes one NADPH (costs ~2.5 ATP), so the total deficit is greater still.
This is why dietary fat composition affects metabolic efficiency - saturated fats yield slightly more ATP per carbon than unsaturated fats when fully oxidized.
Key source: Lippincott Illustrated Reviews: Biochemistry, 8th ed., pp. 547-553 - Beta-oxidation reactions and unsaturated fatty acid oxidation
This is a shared conversation. Sign in to Orris to start your own chat.