Tell me the mechanism of action of paracetamol. It must be 1000% correct
Reading File
Finding Sources
Searching PubMed
paracetamol acetaminophen mechanism of action
Reading File
Finding Sources
Searching PubMed
paracetamol acetaminophen mechanism action analgesic antipyretic
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading Article
PMID: 15662292
Reading File
Reading File
Reading File
Loading Image
I now have a thorough, multi-source body of evidence. Here is the complete, authoritative answer.
Mechanism of Action of Paracetamol (Acetaminophen)
Paracetamol's mechanism of action is notably complex - and, frankly, still not completely resolved despite decades of use. This is not an evasion; it is what the best medical textbooks and reviews consistently state. Here is what is known with confidence, and where uncertainty remains.
1. Primary Mechanism: COX-Dependent Inhibition of Prostaglandin Synthesis
The core pharmacological effect of paracetamol is inhibition of prostaglandin E2 (PGE2) synthesis, which drives both its antipyretic and analgesic actions.
-
Antipyresis: PGE2 is the key mediator that raises the hypothalamic thermoregulatory set point during fever. Paracetamol reduces PGE2 levels in the hypothalamus, lowering the set point back to normal. The antipyretic potency of drugs correlates directly with their ability to inhibit brain cyclooxygenase. - Harrison's Principles of Internal Medicine, 22nd ed.
-
Analgesia: PGE2 sensitizes peripheral nociceptors and central pain pathways. By reducing PGE2 synthesis, paracetamol raises the pain threshold both centrally and, to a limited degree, peripherally.
How it inhibits COX - a critical nuance
Paracetamol is NOT a conventional NSAID. NSAIDs (e.g., ibuprofen, aspirin) bind the COX active site directly. Paracetamol works differently:
- It acts as a reducing co-substrate at the peroxidase (POX) site of COX-1 and COX-2, rather than binding the cyclooxygenase active site. - Firestein & Kelley's Textbook of Rheumatology
- Its inhibitory potency depends heavily on the peroxide tone of the tissue: high-peroxide environments (e.g., activated macrophages, platelets at sites of inflammation) render paracetamol ineffective, which explains why it has no meaningful anti-inflammatory action at therapeutic doses.
- In low-peroxide environments (e.g., CNS, resting vascular endothelial cells), it inhibits COX effectively.
- It also inhibits more when arachidonic acid substrate concentrations are low (<5 µmol/L). At low AA levels, PGs are made predominantly via COX-2; this may explain why paracetamol behaves pharmacologically like a "selective COX-2 inhibitor" despite not binding the COX-2 active site. - Graham & Scott, Am J Ther, 2005 [PMID: 15662292]
Why it has no anti-inflammatory activity
- Inflammation markedly upregulates COX-2 expression, increasing enzyme concentration at the site. Because paracetamol's inhibitory potency is inversely related to COX enzyme concentration (unlike NSAIDs which simply occupy the active site), it becomes ineffective against high COX-2 expression at inflammatory sites. - Firestein & Kelley's Textbook of Rheumatology
- Paracetamol is inactivated peripherally and is active primarily in the CNS, which is why it maintains antipyresis and central analgesia but not peripheral anti-inflammatory effects. - Lippincott Illustrated Reviews: Pharmacology
2. COX-3: The Central Splice Variant Hypothesis
In the brain, paracetamol may preferentially inhibit COX-3, a splice variant of COX-1 that retains intron-1 sequence and is:
- Highly expressed in the cerebral cortex and heart
- More sensitive to inhibition by paracetamol than COX-1 or COX-2
- Not found outside the CNS
This is why paracetamol in the brain (after P450-mediated oxidation) can inhibit COX activity and reduce PGE2 even though it is relatively ineffective peripherally. - Harrison's Principles of Internal Medicine; Firestein & Kelley's
Important caveat: More recent studies have questioned whether COX-3 is clinically relevant as the primary mechanism, since genomic and kinetic analysis indicates it is unlikely to fully explain paracetamol's effects. COX-3 does not arise from a unique gene and is a variant of COX-1. - Firestein & Kelley's Textbook of Rheumatology
3. Endocannabinoid / AM404 Pathway
A major and increasingly well-supported mechanism involves a metabolite of paracetamol formed in the CNS:
- In the brain, paracetamol is deacetylated to p-aminophenol, which then conjugates with arachidonic acid to form AM404 (N-arachidonoylaminophenol) - a potent endocannabinoid.
- AM404 activates CB1 cannabinoid receptors and TRPV1 (transient receptor potential vanilloid 1) channels, both of which contribute to analgesia.
- AM404 also inhibits reuptake of endocannabinoids (particularly anandamide), amplifying the endocannabinoid signal.
- AM404 independently reduces COX-1 expression and PGE2 production via mechanisms not dependent on TRPV1 signaling. - Firestein & Kelley's Textbook of Rheumatology
4. Activation of Descending Serotonergic Pain Inhibitory Pathways
There is considerable evidence that paracetamol's analgesic effect is centrally mediated through:
- Activation of descending serotonergic (5-HT) inhibitory pathways from the brainstem (periaqueductal gray and raphe nuclei) to the dorsal horn of the spinal cord
- This reduces the transmission of pain signals in the spinal cord
- The primary trigger for this activation may still be inhibition of PG synthesis centrally, but the downstream mechanism involves serotonin. - Graham & Scott, Am J Ther [PMID: 15662292]
5. TRPA1 Activation by NAPQI
The toxic metabolite NAPQI (N-acetyl-p-benzoquinone imine), at sub-toxic concentrations, may activate TRPA1 channels, contributing to analgesic effects. However, this metabolite is rapidly detoxified by glutathione under therapeutic doses, and its analgesic contribution at normal dosing is unclear. - Firestein & Kelley's Textbook of Rheumatology
Summary Table
| Mechanism | Effect | Confidence Level |
|---|---|---|
| COX inhibition via peroxidase site (low peroxide/low AA environments) | Antipyresis + Analgesia | Well-established |
| COX-3 inhibition in CNS | Antipyresis + Central Analgesia | Supported but questioned |
| AM404 → CB1 + TRPV1 activation | Central Analgesia | Increasingly supported |
| Descending serotonergic pathway activation | Central Analgesia | Good evidence |
| NAPQI → TRPA1 activation | Possible analgesic contribution | Preliminary |
Metabolism Pathway (Relevant to Mechanism)
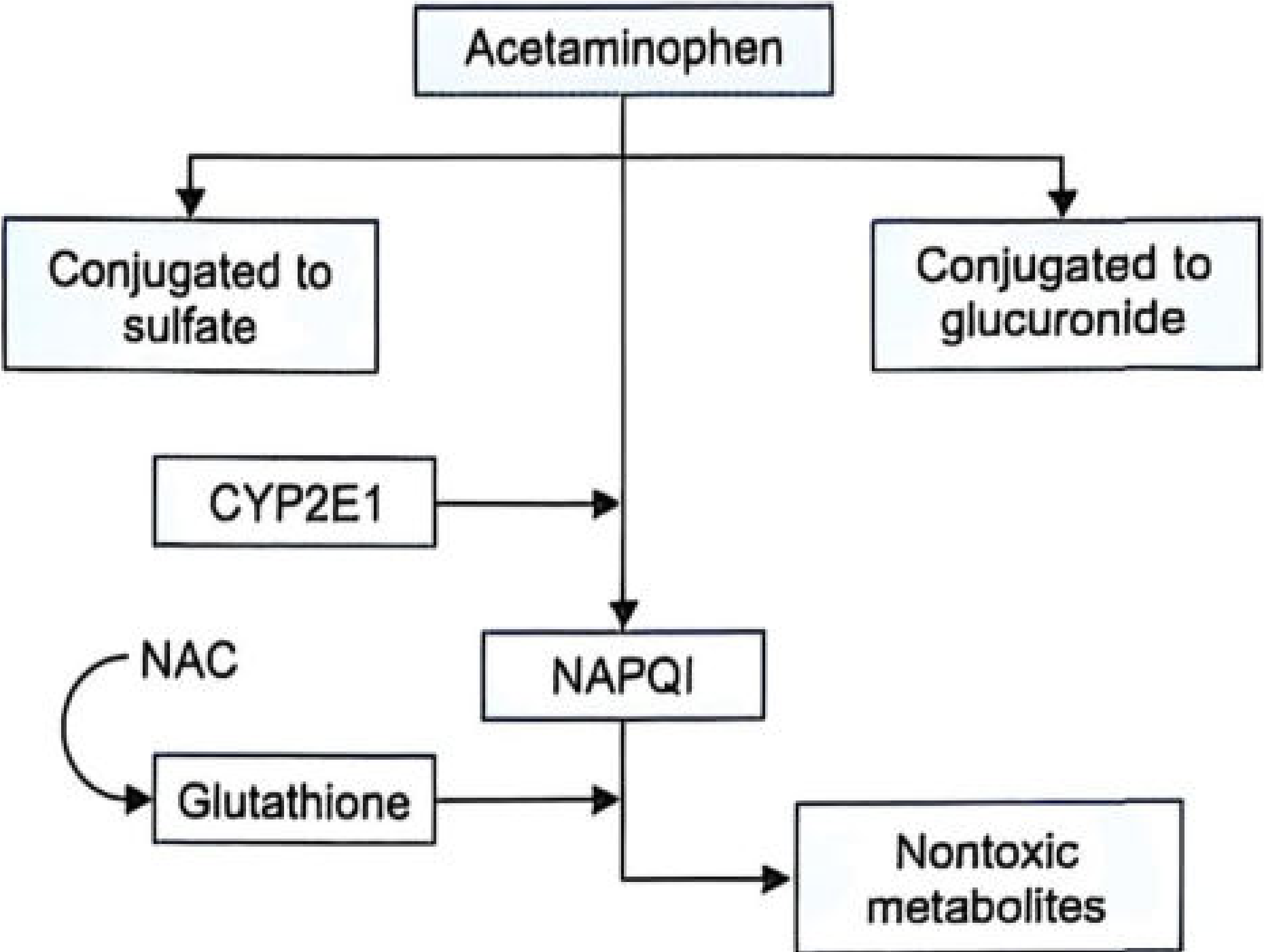
- ~85-90% of paracetamol undergoes glucuronidation and sulfation → nontoxic urinary excretion
- ~5-10% is oxidized by CYP2E1 (minor: CYP1A2, CYP3A4) → NAPQI
- Under therapeutic doses, NAPQI is rapidly conjugated with glutathione → excreted safely
- In overdose, glutathione stores are exhausted → NAPQI accumulates → covalent binding to hepatic proteins → centrilobular necrosis
- Rosen's Emergency Medicine; The Essentials of Forensic Medicine & Toxicology, 36th ed.
Key Distinctions from NSAIDs
| Property | Paracetamol | NSAIDs (e.g., ibuprofen) |
|---|---|---|
| Analgesic | Yes | Yes |
| Antipyretic | Yes | Yes |
| Anti-inflammatory | No | Yes |
| GI toxicity | Minimal | Yes |
| Platelet effect | None | Yes (inhibits) |
| Renal effects | Minimal at therapeutic doses | Yes |
| Cardiovascular risk | Low | Increased (esp. COX-2 inhibitors) |
| Site of COX inhibition | Primarily CNS | Peripheral + CNS |
Sources:
- Harrison's Principles of Internal Medicine, 22nd ed. - "Mechanisms of Antipyretic Agents"
- Firestein & Kelley's Textbook of Rheumatology - "Mechanism of Acetaminophen"
- Lippincott Illustrated Reviews: Pharmacology - COX inhibition, acetaminophen vs. ibuprofen
- Rosen's Emergency Medicine - Acetaminophen pharmacology and toxicology
- The Essentials of Forensic Medicine & Toxicology, 36th ed. - Metabolism pathway
- Graham GG & Scott KF, Mechanism of action of paracetamol, Am J Ther, 2005 - foundational review
This is a shared conversation. Sign in to Orris to start your own chat.