Pathoanatomy kidney pathology
Reading File
Finding Sources
Finding Sources
Asking for Preferences
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Searching Images
kidney glomerulonephritis histology light microscopy
Searching Images
acute tubular necrosis ATN histology pyelonephritis interstitial nephritis
Here is a comprehensive overview of kidney pathoanatomy, covering all four major compartments with detailed histology and mechanisms.
Kidney Pathoanatomy — Comprehensive Overview
Biopsy Technique & Interpretation Framework
A kidney biopsy is processed with three complementary modalities:
- Light microscopy (LM): H&E (cellularity/architecture), PAS (carbohydrate moieties in membranes), Jones-methenamine silver (basement membrane structure), Congo red (amyloid), Masson's trichrome (collagen/fibrosis)
- Immunofluorescence (IF): IgG, IgM, IgA, C1q, C3; pattern is either "lumpy-bumpy" (immune complex deposits) or linear (anti-GBM antibody)
- Electron microscopy (EM): precise location of deposits (subepithelial, subendothelial, mesangial), basement membrane integrity, foot process effacement
Biopsy reporting terms:
- Focal = <50% glomeruli involved; Diffuse = >50%
- Segmental = part of one tuft; Global = entire tuft
- Endocapillary proliferation = cells within capillary loops; Extracapillary = cells in Bowman's space (crescents)
- Crescents = fibrocellular collections in Bowman's space — indicate rapid progression
- Interstitial fibrosis = ominous sign of irreversibility
Harrison's Principles of Internal Medicine 22E, p. 2459–2460
I. GLOMERULAR DISEASES
Classification by Syndrome
| Syndrome | Hallmark | Examples |
|---|---|---|
| Nephritic | Hematuria, RBC casts, hypertension, oliguria, mild proteinuria | PSGN, IgA nephropathy, ANCA vasculitis |
| Nephrotic | Massive proteinuria (>3.5 g/day), hypoalbuminemia, edema, hyperlipidemia | MCD, FSGS, MN, MPGN |
| RPGN | Rapid GFR decline over days–weeks, often crescents | Anti-GBM, ANCA, immune complex |
A. Acute Endocapillary Proliferative GN (Poststreptococcal GN — PSGN)
Prototype of the nephritic syndrome.
Mechanism: Nephritogenic strains of Group A Streptococcus (M-types); key antigens are SPEB (streptococcal pyrogenic exotoxin B) and NAPlr (nephritis-associated plasmin receptor). Circulating immune complexes + alternate complement pathway activation.
Histology (LM):
- Diffuse mesangial + endocapillary hypercellularity
- PMN infiltration of capillary loops
- Granular subendothelial + subepithelial ("hump") immune deposits of IgG, IgM, C3
IF: Granular IgG, IgM, C3 — "starry sky" pattern
EM: Subepithelial electron-dense humps (pathognomonic)
Labs: ↓C3, ↓CH50, normal C4 (alternate pathway); streptozyme test positive in 80–95%
Schematic:
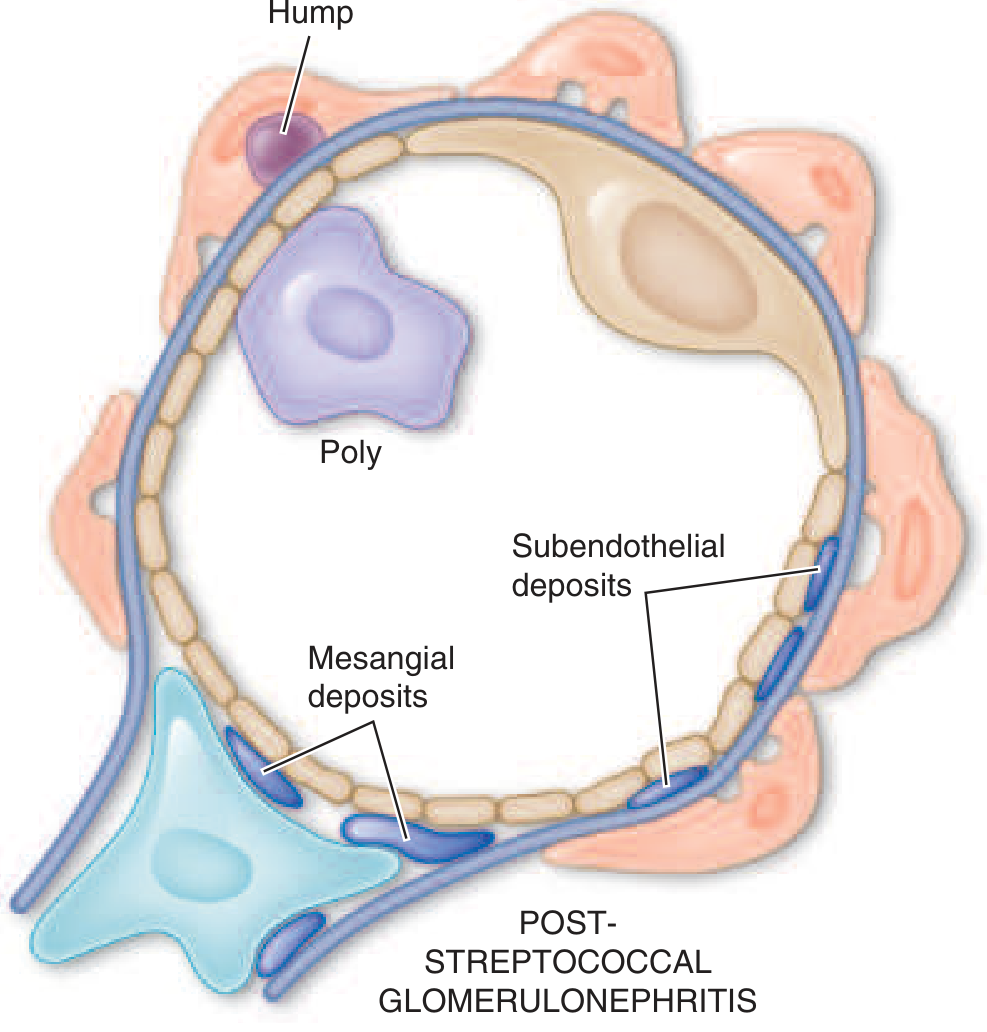
Glomerular Schematic — PSGN showing mesangial deposits and subepithelial humps. — Harrison's 22E
Harrison's Principles of Internal Medicine 22E, p. 2460–2461
B. Anti-GBM Disease / Goodpasture Disease
Mechanism: Autoantibodies against α3(IV)NC1 domain of type IV collagen (the major GBM constituent). The collagen IV network forms a "chicken wire" structure — α3:α4:α5 heterotrimers predominate in GBM.
Histology: Crescentic (extracapillary proliferative) GN; linear IgG along GBM by IF
Clinical: RPGN ± pulmonary hemorrhage (Goodpasture syndrome)
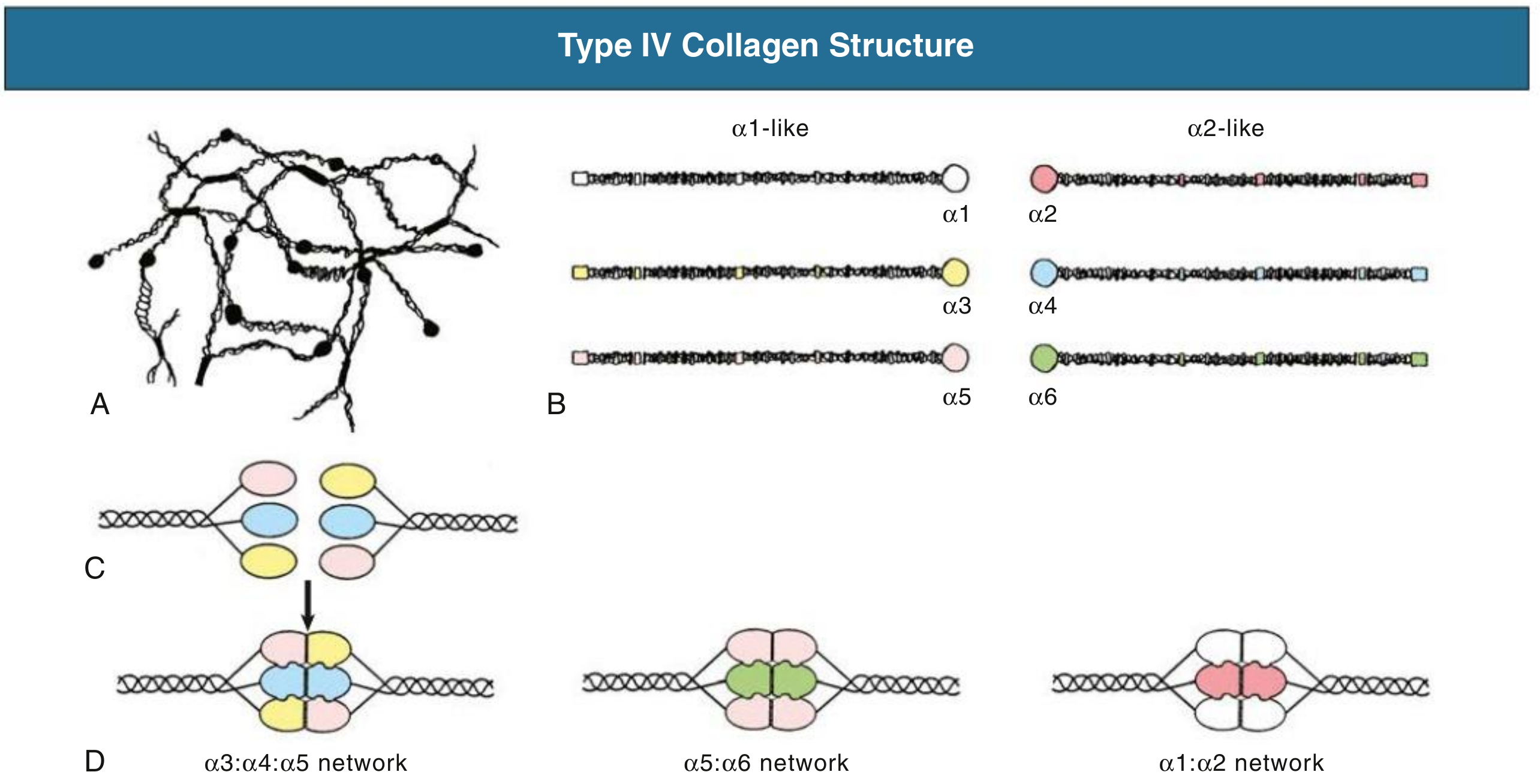
Type IV collagen α-chain networks in GBM — Comprehensive Clinical Nephrology 7e
Comprehensive Clinical Nephrology 7th Ed., p. 345
C. IgA Nephropathy (Berger Disease)
Most common primary GN worldwide. Mechanism: aberrantly glycosylated IgA1 → immune complex formation → mesangial deposition → complement activation.
Histology: Mesangial proliferative GN (most common); may show focal segmental or diffuse proliferative patterns; crescents in aggressive disease
IF: Dominant or co-dominant mesangial IgA + IgG + C3
EM: Electron-dense mesangial deposits
Progression risk: Oxford MEST-C score (Mesangial hypercellularity, Endocapillary hypercellularity, Segmental sclerosis, Tubular atrophy/interstitial fibrosis, Crescents)
D. Membranous Nephropathy (MN)
Prototype of nephrotic syndrome in adults. Primary MN: autoantibodies against PLA2R (phospholipase A2 receptor) in ~70% of cases; also THSD7A, NELL-1, DNAJB9.
Histology (LM):
- Diffuse thickening of GBM without increased cellularity
- "Spike and dome" pattern on Jones silver stain (silver-negative deposits between silver-positive spikes)
IF: Granular IgG + C3 along capillary walls
EM: Subepithelial deposits with overlying GBM spikes ("spikes and domes"); foot process effacement
Stages (Ehrenreich-Churg): I → IV (progressive incorporation of deposits into GBM)
E. Focal Segmental Glomerulosclerosis (FSGS)
Leading cause of nephrotic syndrome in adults (especially Black patients).
Primary FSGS: Circulating permeability factor (podocyte injury). Variants: NOS, Tip, Cellular, Collapsing (worst prognosis — associated with HIV, COVID-19), Perihilar (secondary to hyperfiltration)
APOL1 risk alleles (G1, G2) dramatically increase risk in patients of West African ancestry.
Histology: Focal (<50% glomeruli), segmental sclerosis with hyalinosis; foot process effacement on EM; tubular atrophy proportional to sclerosis
IF: Non-specific IgM, C3 in sclerosed segments (trapped, not immune complex)
F. Membranoproliferative GN (MPGN)
Histology hallmark: Mesangial interposition into GBM → "tram-track" or double-contour appearance on PAS/Jones silver
Two pathogenic mechanisms:
- Immune complex–mediated (IF: IgG, IgM, C3, C4) — HCV-associated cryoglobulinemia, SLE, bacterial infections
- Complement-mediated (IF: C3 dominant, no Ig) — C3GN, Dense Deposit Disease (DDD); associated with C3 nephritic factor, CFH mutations
EM: DDD shows dense osmiophilic deposits within the GBM lamina densa ("sausage-shaped")
G. Lupus Nephritis (ISN/RPS Classification)
| Class | Morphology | Clinical Significance |
|---|---|---|
| I | Normal LM, mesangial deposits by IF/EM | Minimal disease |
| II | Mesangial proliferative | Mild |
| III | Focal proliferative (<50%) | Moderate — treat |
| IV | Diffuse proliferative (≥50%) | Severe — aggressive therapy |
| V | Membranous | Nephrotic syndrome |
| VI | Advanced sclerosis | ESKD |
IF: Full-house pattern — IgG, IgM, IgA, C3, C1q
EM: Subendothelial + mesangial deposits; "fingerprint" deposits in cryoglobulinemia; tubuloreticular inclusions in endothelium (interferon signature)
H. Diabetic Kidney Disease (DKD)
Histology hallmarks (by LM):
- GBM thickening — first lesion identifiable by EM (within 2 years of onset)
- Mesangial expansion — first lesion by LM (PAS stain; 4–5 years after T1DM onset)
- Nodular glomerulosclerosis (Kimmelstiel-Wilson nodules) — PAS-positive acellular mesangial nodules; pathognomonic
- Arteriolar hyalinosis (both afferent AND efferent — specific for diabetes)
- Tubular basement membrane thickening
IF: Linear GBM staining for IgG (non-immune, trapping)
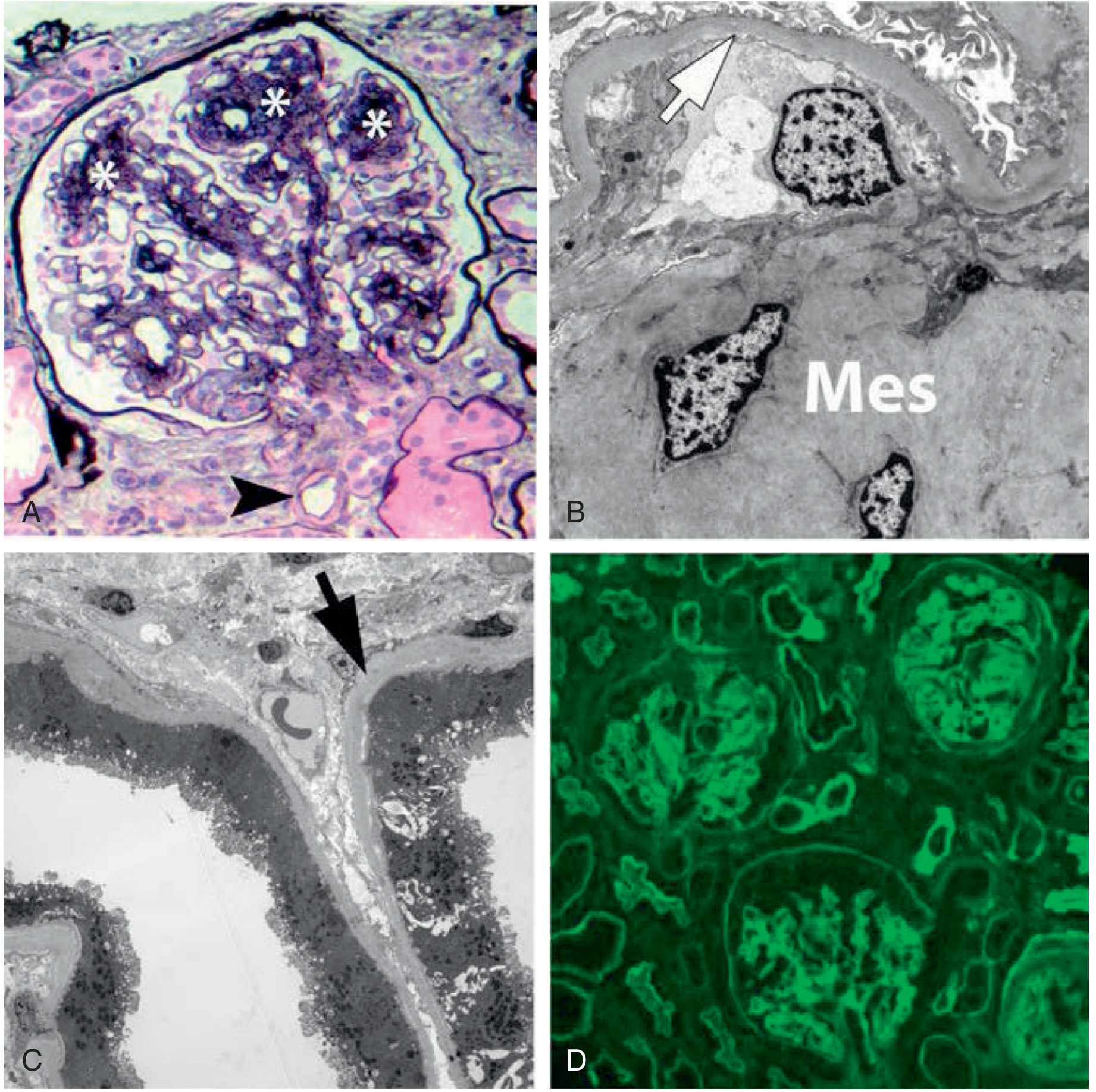
KW nodules (asterisks), arteriolar hyalinosis (arrowhead), Jones silver stain — Brenner & Rector's The Kidney
Brenner and Rector's The Kidney, p. 1778
I. Monoclonal Immunoglobulin Deposition Disease (MIDD)
Includes LCDD (light chain), HCDD (heavy chain), LHCDD.
LM: Nodular mesangial sclerosis mimicking DKD (2/3 of LCDD cases)
Key feature: Ribbon-like eosinophilic deposits along tubular basement membranes (predominantly distal tubules)
IF: Diffuse linear TBM and GBM staining for monotypic κ (LCDD) or a specific heavy chain (HCDD)
EM: Finely granular "powdery" electron-dense deposits along inner GBM and outer TBM
Comprehensive Clinical Nephrology 7e, p. 394
II. TUBULAR & INTERSTITIAL DISEASES
A. Acute Tubular Necrosis (ATN)
Most common cause of intrinsic AKI. Caused by ischemia/reperfusion or nephrotoxins (aminoglycosides, contrast, cisplatin, myoglobin).
Pathophysiology:
- Tubular cell vacuolation → loss of brush border → disruption of epithelial polarity
- Intratubular obstruction by cellular debris
- Backleak of filtrate through denuded epithelium
- Afferent arteriolar vasoconstriction (tubuloglomerular feedback)
- "Trading away" GFR for medullary oxygenation preservation
Site of injury: Outer medulla (most severe) — S3 segment of proximal tubule (ischemia); S1/S2 (aminoglycosides)
Histology:
- Tubular cell vacuolation, brush border loss, epithelial disruption
- Intratubular casts (muddy brown granular casts on urinalysis)
- Focal patchy necrosis (may be missed on cortical biopsy)
- Patchy interstitial mononuclear infiltrates (without tubulitis)
- Evidence of regeneration (mitoses, flattened epithelium) alongside fresh injury
Urinalysis: Muddy brown coarse granular casts; FENa >1%, FEurea >35%
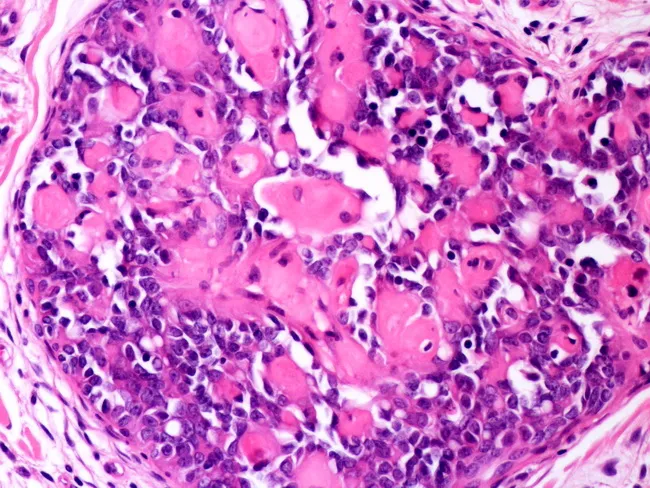
H&E of tubulointerstitium — lymphocytic infiltration, tubulitis, interstitial edema — compatible with AIN/ATN pattern
Comprehensive Clinical Nephrology 7e, p. 990
B. Acute Interstitial Nephritis (AIN)
Accounts for 15–27% of biopsies done for AKI. Most common cause: drugs (antibiotics > PPIs > NSAIDs).
Histology:
- Interstitial inflammatory infiltrate — predominantly T lymphocytes, plasma cells, eosinophils (especially drug-induced)
- Interstitial edema
- Tubulitis — lymphocytes infiltrating tubular epithelium (distinguishes AIN from ATN)
- Tubular atrophy and fibrosis if chronic
IF: May show linear or granular IgG along TBMs (drug-induced)
Clinical triad (classic but uncommon): Fever + rash + eosinophilia (present in <10–30% of drug-induced AIN)
Mean latency: ~10 days after drug exposure; NSAIDs may take months
Comprehensive Clinical Nephrology 7e, p. 990–991
C. Chronic Pyelonephritis / Reflux Nephropathy
Mechanism: Vesicoureteral reflux → ascending bacterial infection → recurrent cortical scarring
Gross pathology: Coarse cortical scars, irregular surface, dilated calyces (clubbing) overlying scars
Histology:
- Interstitial fibrosis + chronic inflammatory infiltrate
- Thyroidization of tubules — dilated tubules packed with pink colloid-like casts resembling thyroid follicles
- Periglomerular fibrosis and global glomerulosclerosis
- Vascular intimal thickening
D. Light Chain Cast Nephropathy (Myeloma Kidney)
Histology: Large, fractured, laminated, bright eosinophilic intratubular casts in distal tubules/collecting ducts; multinucleated giant cell reaction; tubular cell injury
Pathogenesis: Monoclonal free light chains (Bence Jones) overwhelm proximal tubule reabsorption → precipitate with Tamm-Horsfall protein in distal tubule (low pH, high NaCl, diuretics)
III. VASCULAR DISEASES
A. Hypertensive Nephrosclerosis (Benign)
Second most common cause of ESKD after diabetic nephropathy in the US (~27% of ESKD cases).
Histology:
- Arteriolar hyalinosis (arteriolosclerosis) — pink homogeneous material replacing media of afferent arterioles
- Interlobular artery intimal hyperplasia — fibrous thickening ("onion-skin" mild form)
- Ischemic glomerulosclerosis — global sclerosis, wrinkling/retraction of GBM (ischemic collapse)
- Tubular atrophy, interstitial fibrosis
- No immune deposits (key distinguishing feature)
Genetic risk: APOL1 G1/G2 alleles markedly increase ESKD risk in African Americans
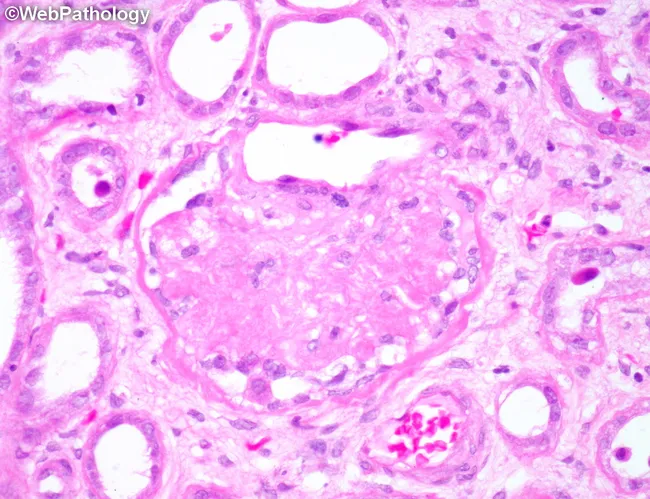
Globally sclerotic glomerulus, arteriolar hyalinosis, tubular atrophy — chronic nephrosclerosis. H&E.
Harrison's 22E, p. 2462
B. Malignant Hypertension (Malignant Nephrosclerosis)
Mechanism: Extreme hemodynamic stress → endothelial injury → fibrinoid necrosis of vessel walls
Histology:
- Fibrinoid necrosis of arterioles (MSB stain: fibrin = red)
- Onion-skin lesion (hyperplastic arteriolosclerosis) — concentric laminated smooth muscle + collagen proliferation in interlobular arteries; markedly narrows lumen
- Thrombotic microangiopathy (TMA) — fibrin thrombi in glomerular capillaries, fragmented RBCs (schistocytes)
- Acute glomerular capillary injury → FSGS-like injury
Labs: Microangiopathic hemolytic anemia (MAHA), thrombocytopenia, AKI
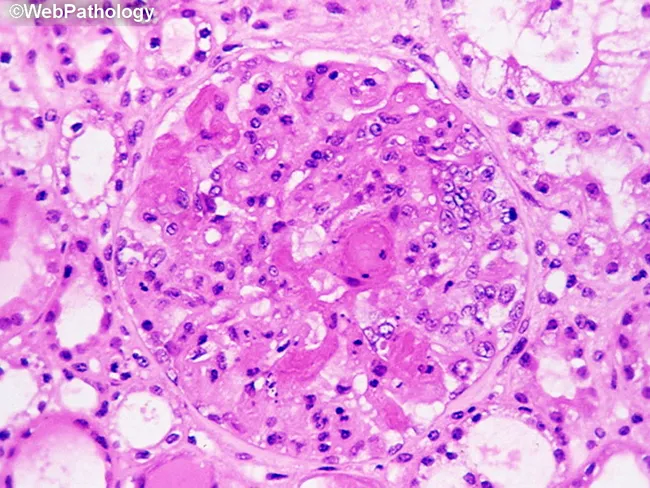
Fibrin deposition in glomerular capillary loops (MSB stain) — malignant nephrosclerosis. Compatible with TMA morphology.
C. Thrombotic Microangiopathy (TMA)
Causes: HUS (STEC-O157:H7 → Shiga toxin → endothelial injury), TTP (ADAMTS13 deficiency), complement-mediated (atypical HUS — CFH/CFI/C3/MCP mutations), malignant hypertension, antiphospholipid syndrome, scleroderma renal crisis
Histology:
- Glomerular capillary fibrin/platelet thrombi
- Endothelial swelling with "bloodless" glomeruli
- Mesangiolysis (dissolution of mesangial matrix)
- Arteriolar intimal mucoid edema, intraluminal thrombi
D. Cholesterol Emboli (Atheroembolism)
Mechanism: Cholesterol crystals released from atheromatous plaques (post-procedure/spontaneous) → lodge in arcuate/interlobular arteries → ischemic damage + local eosinophilic inflammation
Histology:
- Biconvex clefts in vessels (cholesterol dissolved during tissue processing)
- Surrounding giant cell reaction and eosinophilia
- Downstream cortical ischemia/infarction
E. Renal Artery Stenosis
Causes: Atherosclerosis (older, males, proximal RAS) or fibromuscular dysplasia (young women, distal/medial RAS → "string of beads" angiography)
Histology: Ischemic nephropathy — tubular atrophy, arteriolar collapse, global glomerulosclerosis, preserved cortical width (bilateral RAS) or focal cortical thinning (unilateral)
IV. INHERITED / STRUCTURAL / OTHER
A. Alport Syndrome
Mutation in COL4A3, COL4A4 (autosomal) or COL4A5 (X-linked, most common). Leads to absence of α3:α4:α5 type IV collagen network from GBM.
EM (pathognomonic): Irregular thinning and thickening, splitting/lamellation of GBM — "basket-weave" or "moth-eaten" appearance
IF: Absent staining for α3/α4/α5(IV) collagen in GBM (diagnostic)
Clinical: Hematuria → progressive CKD + sensorineural hearing loss + ocular defects (anterior lenticonus)
B. Thin Basement Membrane Disease (Benign Familial Hematuria)
Heterozygous COL4A3/A4 mutations. Diffuse uniform thinning of GBM (<150 nm; normal 300–400 nm) on EM. LM and IF: normal. Benign persistent microscopic hematuria.
C. Polycystic Kidney Disease (PKD)
| ADPKD | ARPKD | |
|---|---|---|
| Gene | PKD1 (85%), PKD2 | PKHD1 |
| Cysts | All nephron segments | Collecting ducts + bile ducts |
| Age | Adult (3rd–5th decade) | Perinatal/childhood |
Pathology: Massively enlarged kidneys with innumerable fluid-filled cysts of all sizes; cysts lined by flattened epithelium; interstitial fibrosis between cysts
Summary Table
| Category | Key Lesion | LM Pattern | IF | EM |
|---|---|---|---|---|
| PSGN | Endocapillary proliferative | PMN infiltration, hypercellularity | Granular IgG, IgM, C3 | Subepithelial humps |
| Anti-GBM | Crescentic GN | Extracapillary proliferation | Linear IgG | GBM disruption |
| IgA nephropathy | Mesangial proliferative | Mesangial expansion | Dominant IgA (mesangial) | Mesangial deposits |
| MN | No proliferation | Thick GBM, spikes | Granular IgG (subepithelial) | Subepithelial deposits |
| FSGS | Focal segmental sclerosis | Segmental hyalinosis | Non-specific IgM/C3 | Foot process effacement |
| MPGN | Lobular, double contours | Tram-track | C3 ± Ig | Subendothelial/intramembranous |
| DKD | Nodular glomerulosclerosis | KW nodules, arteriolar hyalinosis | Linear IgG (non-immune) | GBM thickening, mesangial expansion |
| ATN | Tubular injury | Brush border loss, casts | — | Mitochondrial swelling |
| AIN | Interstitial inflammation | Tubulitis, eosinophils | ± Linear TBM IgG | Normal glomeruli |
| Benign HTN | Arteriosclerosis | Arteriolar hyalinosis, ischemic GS | None | — |
| Malignant HTN | Fibrinoid necrosis, TMA | Onion-skin, thrombi | Fibrin | Endothelial swelling |
| Alport | GBM splitting | Normal LM | Absent α3/α5(IV) | Basket-weave GBM |
Key References:
- Harrison's Principles of Internal Medicine 22E (2025), pp. 2459–2463
- Comprehensive Clinical Nephrology 7th Ed., pp. 344–346, 990–991
- Brenner and Rector's The Kidney, pp. 1778–1780
This is a shared conversation. Sign in to Orris to start your own chat.