Emphysema, it’s types, etiopathogenesis, morphology, and clinical course
Reading File
Finding Sources
Searching PubMed
emphysema pathogenesis types COPD
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
Loading Image
Finding Sources
Reading File
I now have all the content needed for a comprehensive answer. Let me compile the full response.
Emphysema
Definition
Emphysema is defined as the irreversible enlargement of airspaces distal to the terminal bronchiole, accompanied by destruction of alveolar walls, without significant fibrosis. It is classified anatomically (based on which part of the acinus is involved), unlike chronic bronchitis which is defined clinically.
The acinus - the fundamental respiratory unit - consists of the respiratory bronchiole, alveolar ducts, and alveolar sacs. A cluster of 3-5 acini forms a pulmonary lobule.
Types of Emphysema
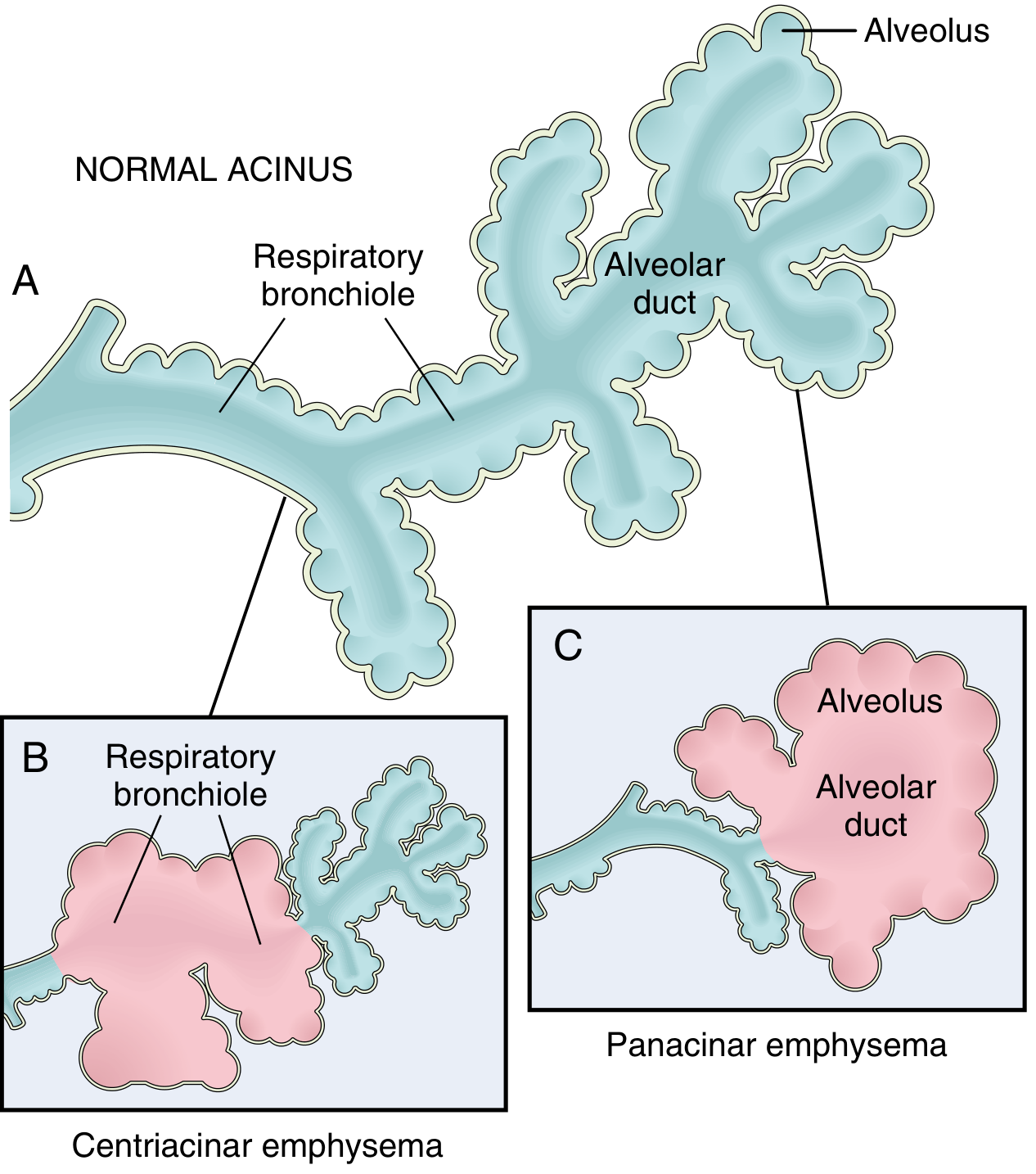
Fig. Clinically significant patterns of emphysema. (A) Normal acinus structure showing respiratory bronchioles, alveolar ducts, and alveoli. (B) Centriacinar emphysema - dilation initially affects the respiratory bronchioles. (C) Panacinar emphysema - initial distention of alveolus and alveolar duct.
There are four major types, of which only the first two cause clinically significant airflow obstruction:
1. Centriacinar (Centrilobular) Emphysema
- Most common form - constitutes >95% of clinically significant cases
- The central/proximal parts of the acinus (respiratory bronchioles) are affected; distal alveoli are spared
- Both emphysematous and normal airspaces coexist within the same acinus and lobule
- Distribution: upper lobes, particularly apical segments
- Etiology: almost exclusively in heavy smokers; often accompanied by chronic bronchitis
- In severe cases, the distal acinus is also affected, making differentiation from panacinar emphysema difficult
2. Panacinar (Panlobular) Emphysema
- The entire acinus is uniformly enlarged - from respiratory bronchiole to terminal alveoli
- Distribution: lower lung zones and anterior margins; most severe at the bases
- Etiology: strongly associated with α1-antitrypsin (AAT) deficiency; exacerbated by smoking
- About 1% of emphysema patients have this defect; >80% of homozygous ZZ individuals develop symptomatic panacinar emphysema, at an earlier age if they smoke
3. Distal Acinar (Paraseptal) Emphysema
- The proximal acinus is normal; the distal portion (alveolar ducts and sacs) is predominantly involved
- More striking adjacent to the pleura, along lobular connective tissue septa, and at the margins of lobules
- Characteristic: multiple enlarged airspaces, 0.5 mm to >2.0 cm in diameter, sometimes forming cystic structures and bullae
- Usually more severe in the upper half of the lungs
- Cause is unknown; typically presents in young adults with spontaneous pneumothorax
- Not significantly associated with airflow obstruction
4. Irregular (Paracicatricial) Emphysema
- Acinus is irregularly involved, almost invariably associated with scarring
- Occurs in small foci adjacent to areas of fibrosis, scarring, or atelectasis
- Clinically insignificant in most cases
Additional Descriptive Forms (not primary anatomical types)
- Bullous emphysema: Large subpleural blebs or bullae (>1 cm diameter) occurring in any form of emphysema, often near the apex; rupture can cause pneumothorax
- Compensatory hyperinflation: Alveolar dilation in response to lung tissue loss elsewhere (e.g., post-lobectomy); not true emphysema as there is no wall destruction
- Obstructive overinflation: Air trapping from subtotal airway obstruction (tumor, foreign body, congenital lobar overinflation); not true emphysema
- Interstitial emphysema: Air entry into connective tissue stroma, mediastinum, or subcutaneous tissue; usually from alveolar tears caused by sudden increases in intraalveolar pressure
Etiopathogenesis
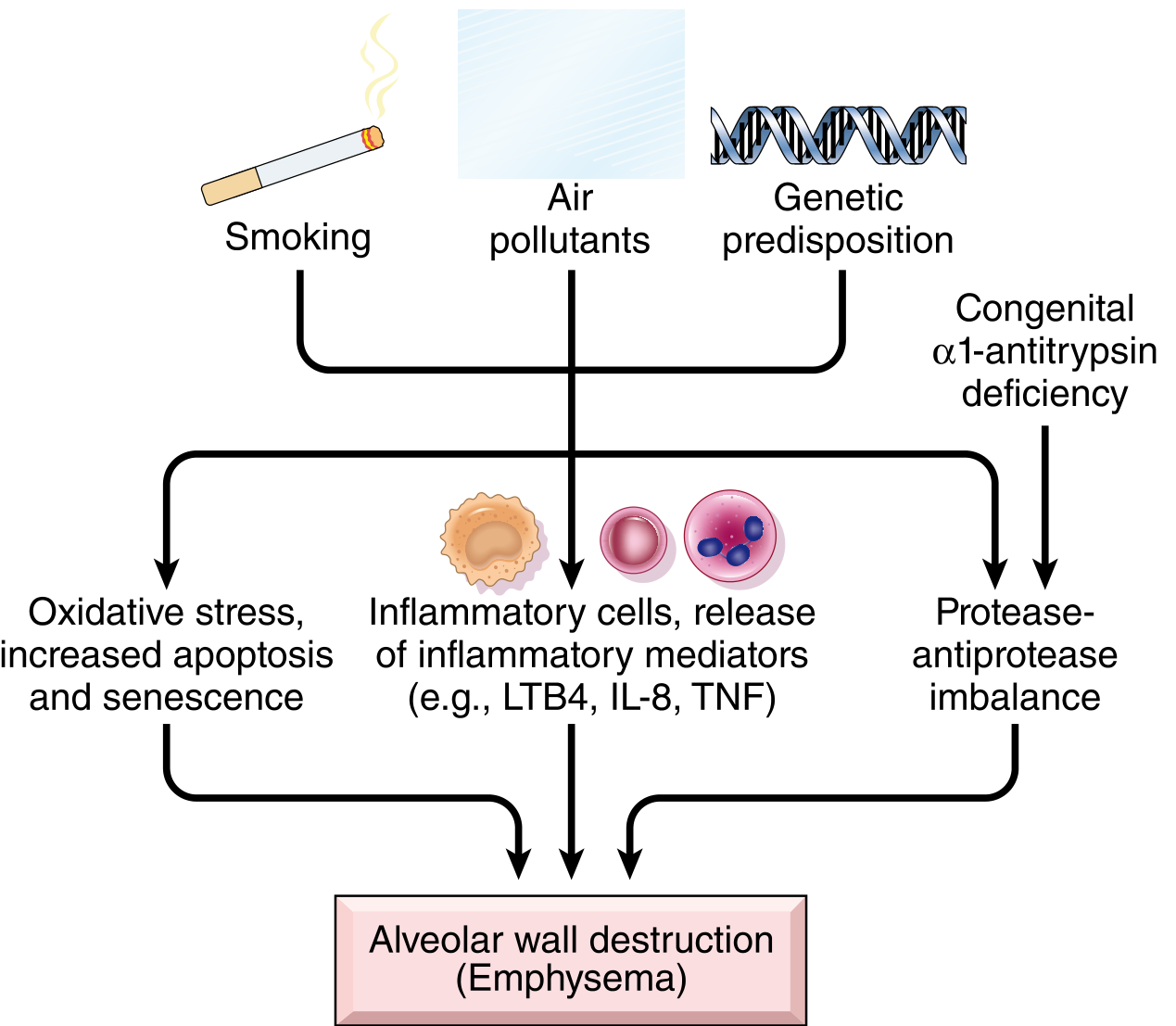
Fig. Pathogenesis of emphysema: three major pathways - oxidative stress/apoptosis/senescence; inflammatory cell recruitment and mediator release (LTB4, IL-8, TNF); and protease-antiprotease imbalance - all converging on alveolar wall destruction.
Clinically significant emphysema is largely confined to smokers and patients with α1-antitrypsin deficiency, highlighting the two cardinal etiologic factors.
1. Toxic Injury and Inflammation
- Inhaled cigarette smoke and noxious particles damage respiratory epithelium, triggering inflammation
- Inflammatory mediators released include leukotriene B4 (LTB4), IL-8, TNF, and others
- Released by resident epithelial cells and macrophages, these mediators:
- Recruit neutrophils, macrophages, CD4+, and CD8+ T cells from the circulation
- Amplify inflammation via proinflammatory cytokines
- Induce structural changes via growth factors
- Bacterial/viral infections do not initiate disease but acutely exacerbate existing disease
2. Protease-Antiprotease Imbalance
- Central mechanism in emphysema pathogenesis
- Proteases (particularly elastase) released from neutrophils and macrophages degrade elastin and other connective tissue components
- In susceptible individuals, there is a relative deficiency of protective antiproteases
- α1-antitrypsin (AAT): the major inhibitor of neutrophil elastase; encoded at the Pi locus on chromosome 14; the ZZ genotype (0.01% of US population) causes markedly decreased serum AAT levels
- Smoking further compounds AAT deficiency by:
- Stimulating recruitment of more neutrophils and macrophages to the lung
- Inactivating AAT via oxidation by cigarette smoke oxidants
- Loss of elastin in alveolar walls removes the radial traction that keeps small airways open, causing respiratory bronchiole collapse during expiration - producing functional airflow obstruction without mechanical obstruction
3. Oxidative Stress
- Tobacco smoke, alveolar damage, and inflammatory cells generate reactive oxygen species (ROS)
- ROS cause tissue damage, endothelial dysfunction, and inflammation
- NRF2 (encoded by NFE2L2) is a transcription factor that senses intracellular oxidants and upregulates cytoprotective genes; mice lacking NRF2 are significantly more sensitive to tobacco smoke
- Genetic variants in NRF2, its regulators, and target genes are linked to smoking-related lung disease in humans
- Oxidants promote apoptosis and cellular senescence of alveolar cells
4. Other Genetic Factors
- Variants of the nicotinic acetylcholine receptor are linked to tobacco addiction behavior, and to both lung cancer and emphysema risk - emphasizing that smoking is the dominant driver
Morphology
Gross Appearance
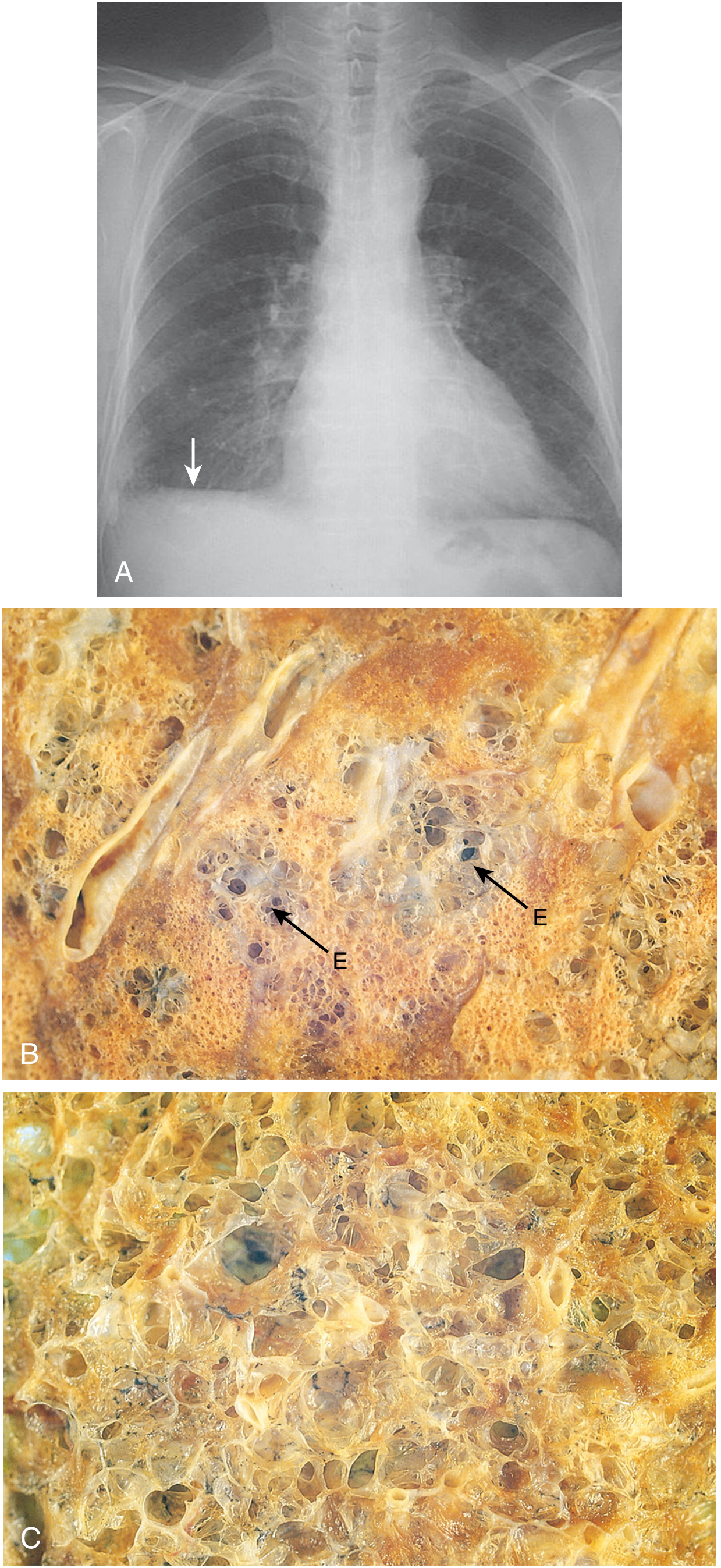
Fig. Emphysema. (A) Chest X-ray showing flattened diaphragm (arrow). (B) Centriacinar emphysema - foci of emphysema (E) surrounded by less affected lung. (C) Panacinar emphysema - diffuse uniform enlargement of all airspaces.
- Advanced emphysema produces voluminous, pale lungs that often overlap the heart anteriorly and flatten the diaphragm
- In smoking-related disease, the upper two-thirds of the lung are more severely affected
- Large alveoli are easily visible on cut sections of fixed lung
- Apical blebs or bullae may be present in advanced disease
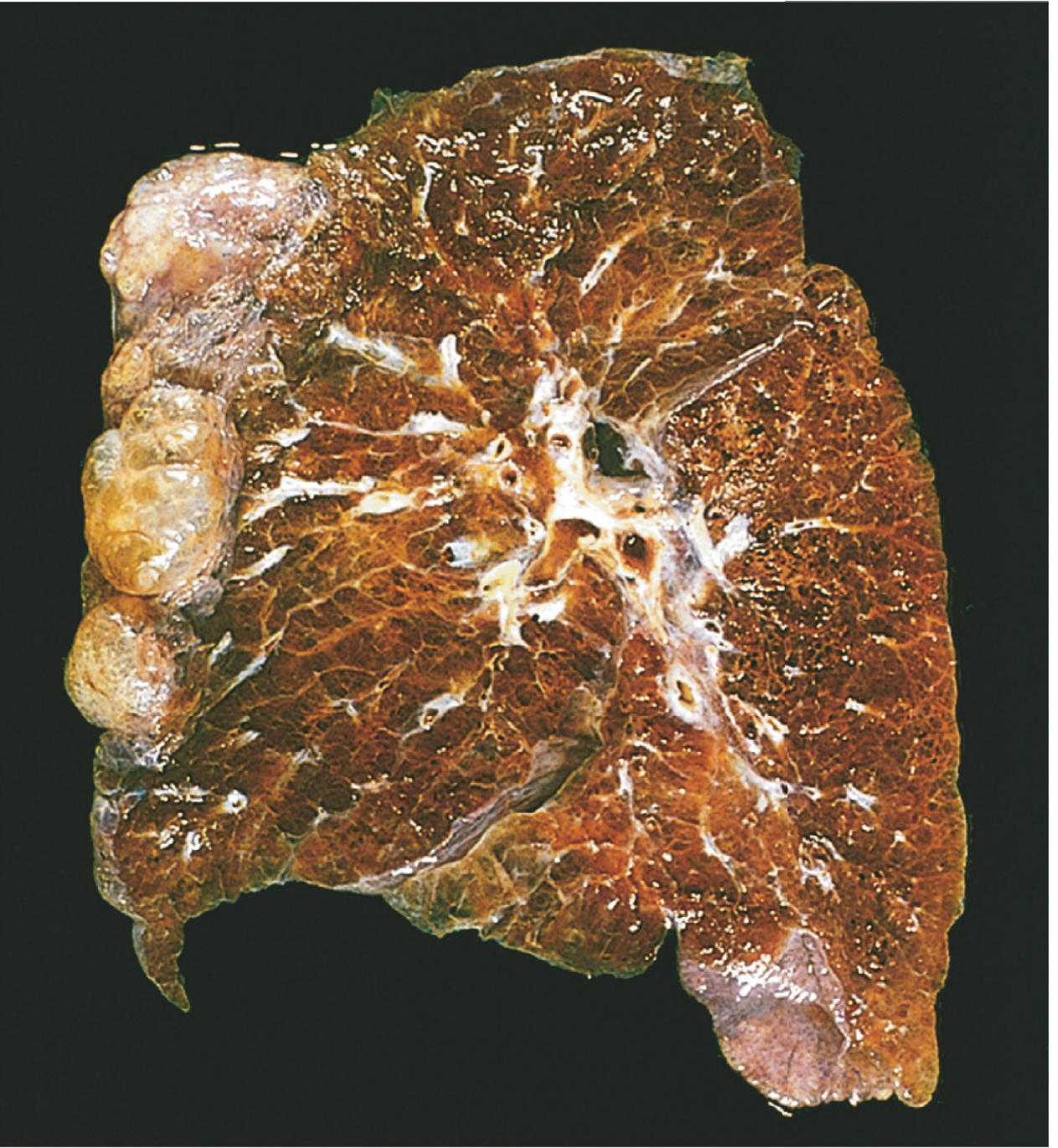
Fig. Bullous emphysema - note the large subpleural bullae at the apex (upper left).
- Panacinar emphysema produces the most impressive gross appearance: paler and more voluminous lungs; lower lobes predominate
- Centriacinar emphysema: lungs are a deeper pink and less voluminous until late stages; upper two-thirds more affected
Microscopic Appearance
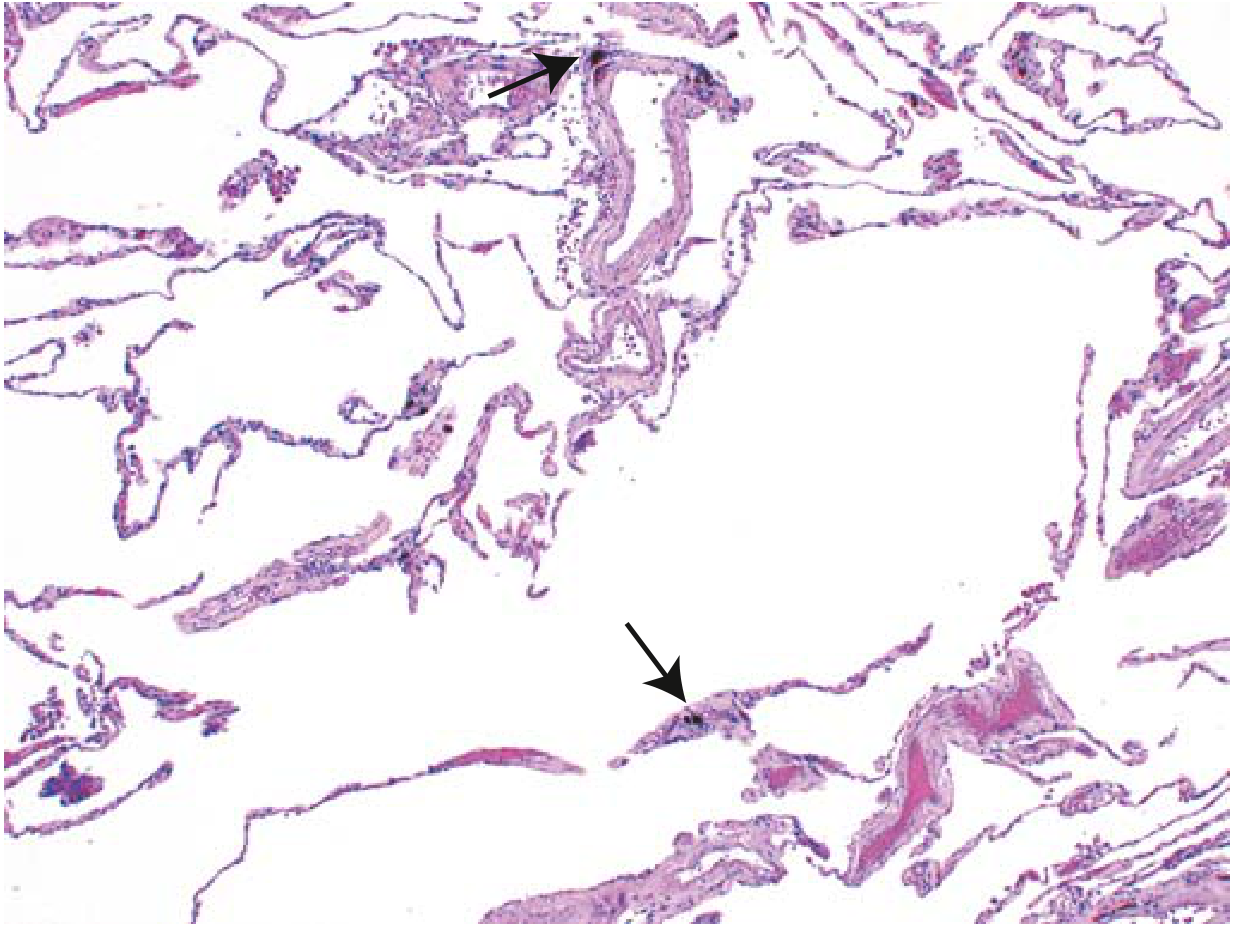
Fig. Pulmonary emphysema (H&E). Markedly enlarged airspaces with destruction of alveolar septa but without fibrosis. Anthracotic pigment visible (arrows).
- Abnormally large alveoli separated by thin septa with focal centriacinar fibrosis
- Loss of attachments between alveoli and small airway walls
- Pores of Kohn become so enlarged that septa appear to "float" or protrude blindly into airspaces with club-shaped ends
- Decrease in capillary bed area due to alveolar wall destruction
- In advanced disease: larger abnormal airspaces, blebs, bullae; deformation and compression of respiratory bronchioles and vasculature
- No significant fibrosis (the key distinguishing histological feature)
- Superimposed inflammatory changes in small airways (related to chronic bronchitis) are common
- Vascular changes of pulmonary hypertension from local hypoxemia and capillary loss
Clinical Course
Presentation
- Most affected patients have a smoking history of ≥40 pack-years
- Onset is insidious: slowly increasing dyspnea on exertion, chronic cough with sputum
- May present with acute exacerbations from superimposed infection
- Key diagnostic test: spirometry showing FEV1/FVC < 0.7
Classic Clinical Phenotypes
| Feature | "Pink Puffer" (Emphysema-dominant) | "Blue Bloater" (Bronchitis-dominant) |
|---|---|---|
| Age | 50-75 years | 40-45 years |
| Build | Thin/cachectic | Overweight |
| Cough | Slight | Prominent, productive |
| Sputum | Scanty | Copious |
| Dyspnea | Severe; progressive | Mild to moderate |
| Cyanosis | Absent/mild | Present (hypoxemia) |
| PaO2 | Near-normal at rest | Reduced |
| PaCO2 | Near-normal | Elevated (hypercapnia) |
| Hematocrit | Normal | Elevated (polycythemia) |
| Cor pulmonale | Uncommon until late | Common |
| Appearance | Barrel chest; hunched over; pursed-lip breathing | Edematous |
Most patients fall somewhere between these two extremes, with a mixture of emphysematous and bronchitic changes.
Mechanisms of Dyspnea and Airflow Obstruction
- Loss of elastic recoil - destruction of elastic tissue in alveolar walls reduces radial traction on surrounding small airways, causing expiratory collapse
- Air trapping - expiratory airway collapse leads to hyperinflation (barrel chest, flattened diaphragm)
- Reduced gas exchange surface - capillary bed destruction lowers diffusion capacity (DLCO)
- Pulmonary hypertension - from hypoxic vasoconstriction and loss of capillary beds; leads to cor pulmonale (right heart failure)
Complications and Prognosis
- Cor pulmonale and right heart failure in advanced disease
- Respiratory failure precipitated by superimposed infection
- Pneumothorax (especially from ruptured bullae in paraseptal emphysema)
- Often fatal from heart failure or respiratory failure from superimposed infection
- Prognosis is significantly worsened by continued smoking
- In AAT-deficient patients, disease occurs earlier, is more severe, and progresses faster, especially in smokers
Sources: Robbins, Cotran & Kumar - Pathologic Basis of Disease (10th ed.), pp. 634-638; Robbins & Kumar Basic Pathology (11th ed.), pp. 447-449
This is a shared conversation. Sign in to Orris to start your own chat.