Teach Cirrhosis according to pathology
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
Loading Image
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
I now have all the material needed for a comprehensive, image-rich pathology lecture on cirrhosis. Let me compile the complete response:
Cirrhosis - A Pathology-Based Teaching Guide
Sources: Robbins & Cotran Pathologic Basis of Disease (10e), Robbins & Kumar Basic Pathology, Goldman-Cecil Medicine, Schwartz's Principles of Surgery
1. Definition
Cirrhosis is defined as the diffuse transformation of the entire liver into regenerative parenchymal nodules surrounded by fibrous bands, with a variable degree of vascular (portosystemic) shunting. It represents the morphologic endpoint of virtually any chronic liver injury.
Three cardinal features must ALL be present:
- Diffuse fibrosis (fibrous septa bridging portal tracts and/or central veins)
- Regenerative nodules (islands of hepatocytes attempting repair)
- Disruption of the normal hepatic architecture (loss of the lobular and vascular organization)
Note: Cirrhosis ≠ Chronic liver failure. Not all cirrhosis leads to liver failure, and not all end-stage liver disease is cirrhotic. (e.g., primary biliary cholangitis, primary sclerosing cholangitis, and nodular regenerative hyperplasia may reach end-stage without fully established cirrhosis.) - Robbins & Cotran PBD
2. Etiology
| Category | Examples |
|---|---|
| Alcohol-associated | Most common in Western nations |
| Viral hepatitis | HBV, HCV, HDV (superinfection) |
| Metabolic | MASLD/NASH (now the most common worldwide), NAFLD |
| Cholestatic/Autoimmune | Primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), autoimmune hepatitis |
| Genetic/Metabolic disorders | Hemochromatosis, Wilson disease, alpha-1-antitrypsin deficiency, glycogen storage diseases, galactosemia, tyrosinemia |
| Vascular | Budd-Chiari syndrome, right heart failure (cardiac cirrhosis) |
| Drugs/Toxins | Methotrexate, amiodarone |
| Cryptogenic | No identifiable cause (~10-15%) |
3. Pathogenesis - The Core Mechanism
The fundamental sequence is: Hepatocyte injury → Inflammation → Stellate cell activation → Fibrosis → Architectural distortion → Nodular regeneration
Step 1: The Key Cell - Hepatic Stellate Cells (Ito Cells)
The central pathogenic feature is activation of hepatic stellate cells (HSCs), also known as Ito cells or perisinusoidal cells. - Goldman-Cecil Medicine
- Normal state: HSCs reside quiescent in the Space of Disse (between hepatocytes and sinusoidal endothelial cells), storing retinoids (vitamin A) as lipid droplets.
- Activated state: In response to chronic liver injury, HSCs undergo a dramatic transformation:
- Lose their vitamin A lipid droplets
- Proliferate and develop prominent rough endoplasmic reticulum
- Transform into contractile myofibroblasts (smooth muscle actin-positive)
- Begin secreting extracellular matrix: collagen types I and III, sulfated proteoglycans, and glycoproteins
Step 2: Triggers of Stellate Cell Activation
Stellate cells are activated by signals from multiple sources:
- Kupffer cells (resident macrophages) release TGF-β, TNF-α, IL-1
- Damaged hepatocytes release reactive oxygen species (ROS), lipid peroxidation products
- Inflammatory cells (T cells, NK cells, NKT cells) release inflammatory cytokines
- TGF-β1 is the dominant pro-fibrotic cytokine
- PDGF (platelet-derived growth factor) is the main stimulus for stellate cell proliferation
Step 3: Progressive Fibrosis
Continued activation leads to:
- Deposition of collagen in the perisinusoidal space (normally collagen-free)
- Formation of fibrous septa linking portal tracts to each other (portal-portal bridging) or to central veins (portal-central bridging)
- Capillarization of sinusoids - loss of sinusoidal fenestrae and deposition of basement membrane → impairs exchange between blood and hepatocytes
- Ductular reactions - proliferating bile ductule-like structures appear, driven by progenitor cell activation; increase with disease progression and are most prominent in cirrhosis
Step 4: Nodular Regeneration
Hepatocytes that survive attempt regeneration, but because the fibrous framework is disrupted:
- Regenerating hepatocytes form discrete nodules (not lobules)
- The regenerative nodules have no proper central vein or portal tract - architecture is completely disorganized
- Adjacent nodules may coalesce over time
Fibrosis Regression (Important Concept)
Fibrosis - and even established cirrhosis - can regress if the underlying cause is eliminated (e.g., alcohol abstinence, cure of hepatitis C with antivirals). Scars become thinner and more densely compacted, then begin to fragment. Fibrous septa break apart and adjacent nodules coalesce into larger islands. - Robbins & Cotran PBD
4. Morphology
Gross Pathology
Fig. 18.6 - Cirrhosis from chronic viral hepatitis. The normally smooth liver capsule is converted into a bumpy, nodular surface with depressed areas of dense scar and bulging regenerative nodules.
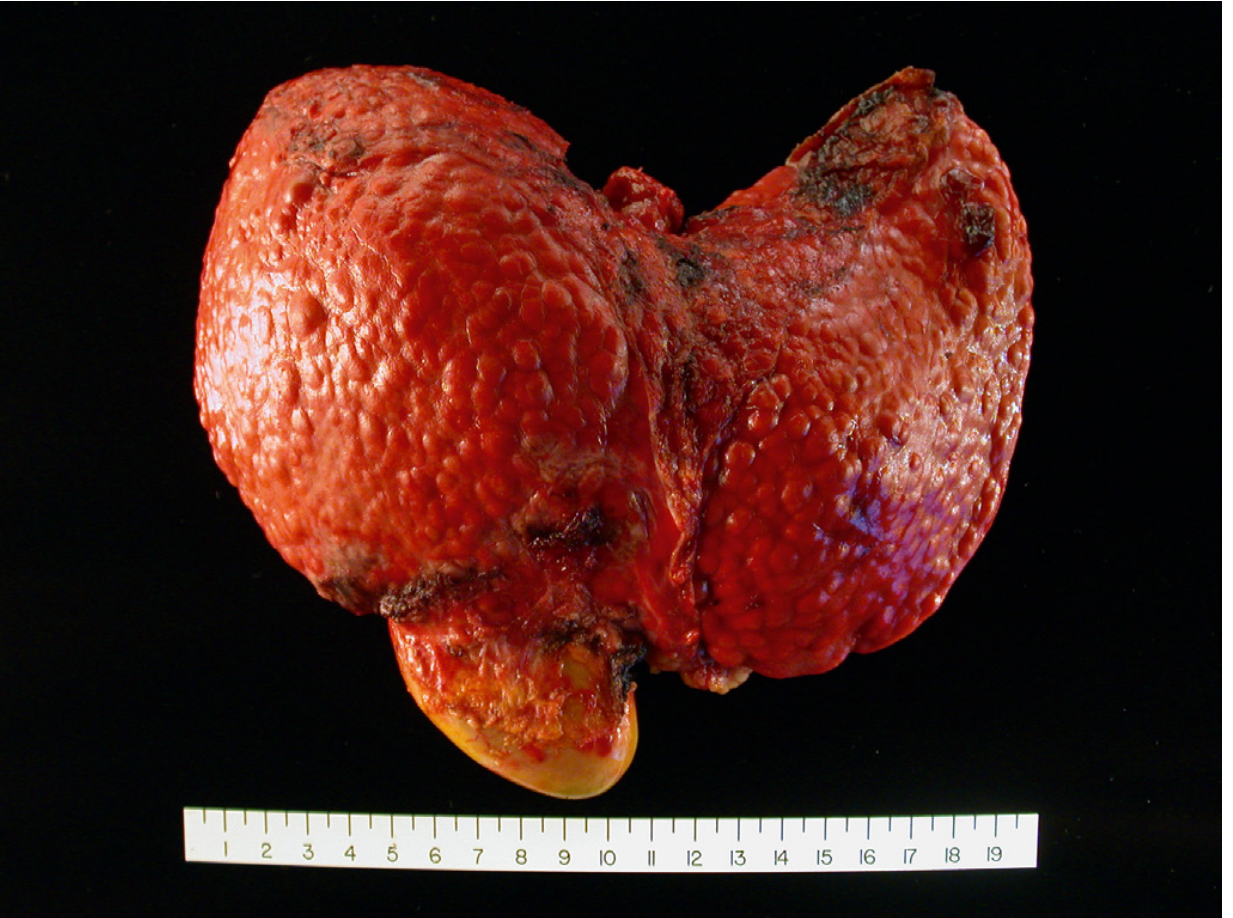
Robbins & Cotran Pathologic Basis of Disease - Gross specimen of cirrhosis from chronic viral hepatitis
Normal vs. Cirrhotic liver comparison (Goldman-Cecil Medicine):
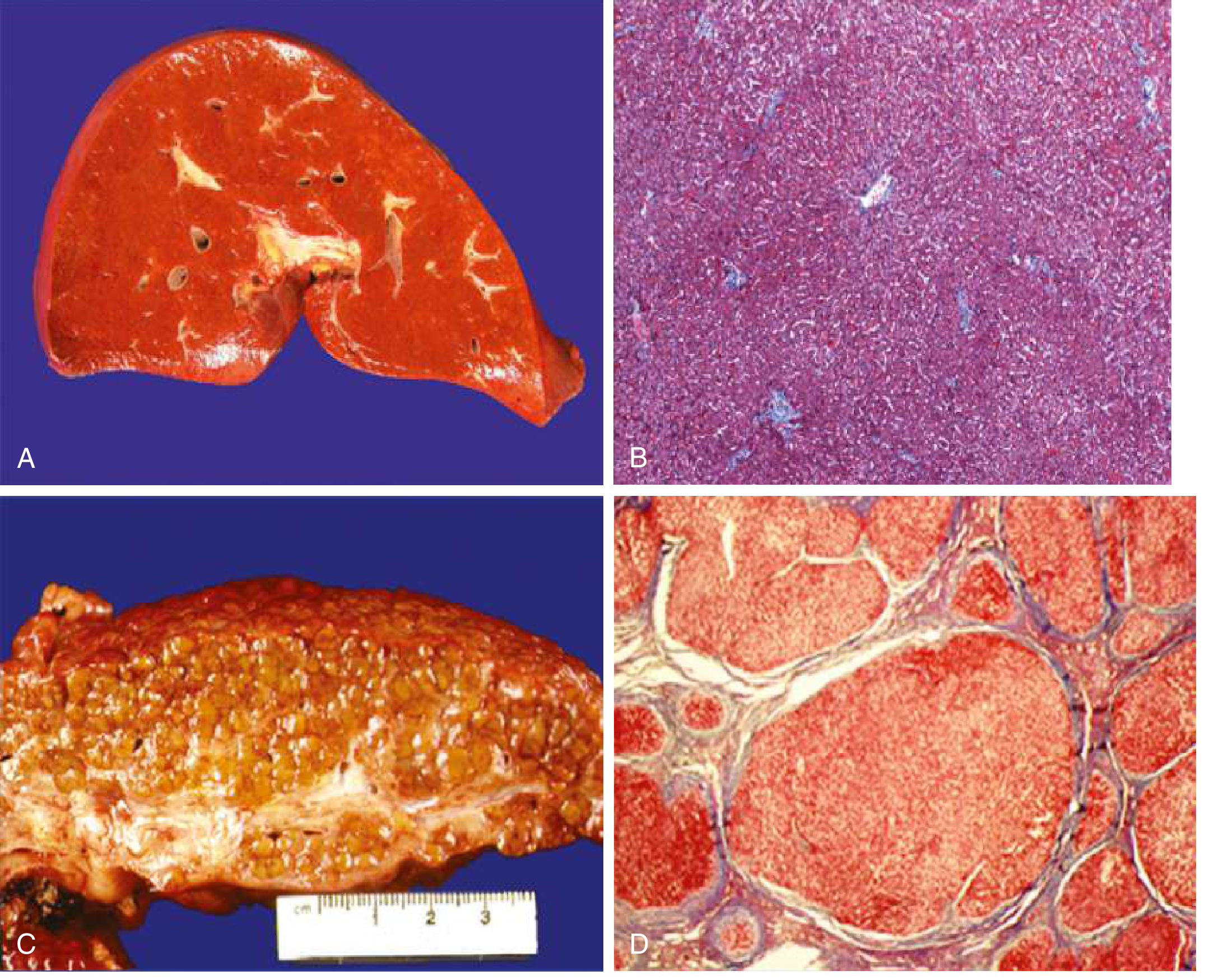
- A: Normal liver - smooth surface, homogeneous texture
- B: Normal histology - organized sinusoids, normal vascular structures
- C: Cirrhotic liver - orange-tawny color, irregular nodular surface
- D: Cirrhotic histology - disorganized architecture, regenerative nodules surrounded by fibrous tissue
Microscopic Pathology
Masson Trichrome stain is the key special stain - highlights collagen (blue) against pink hepatocytes.
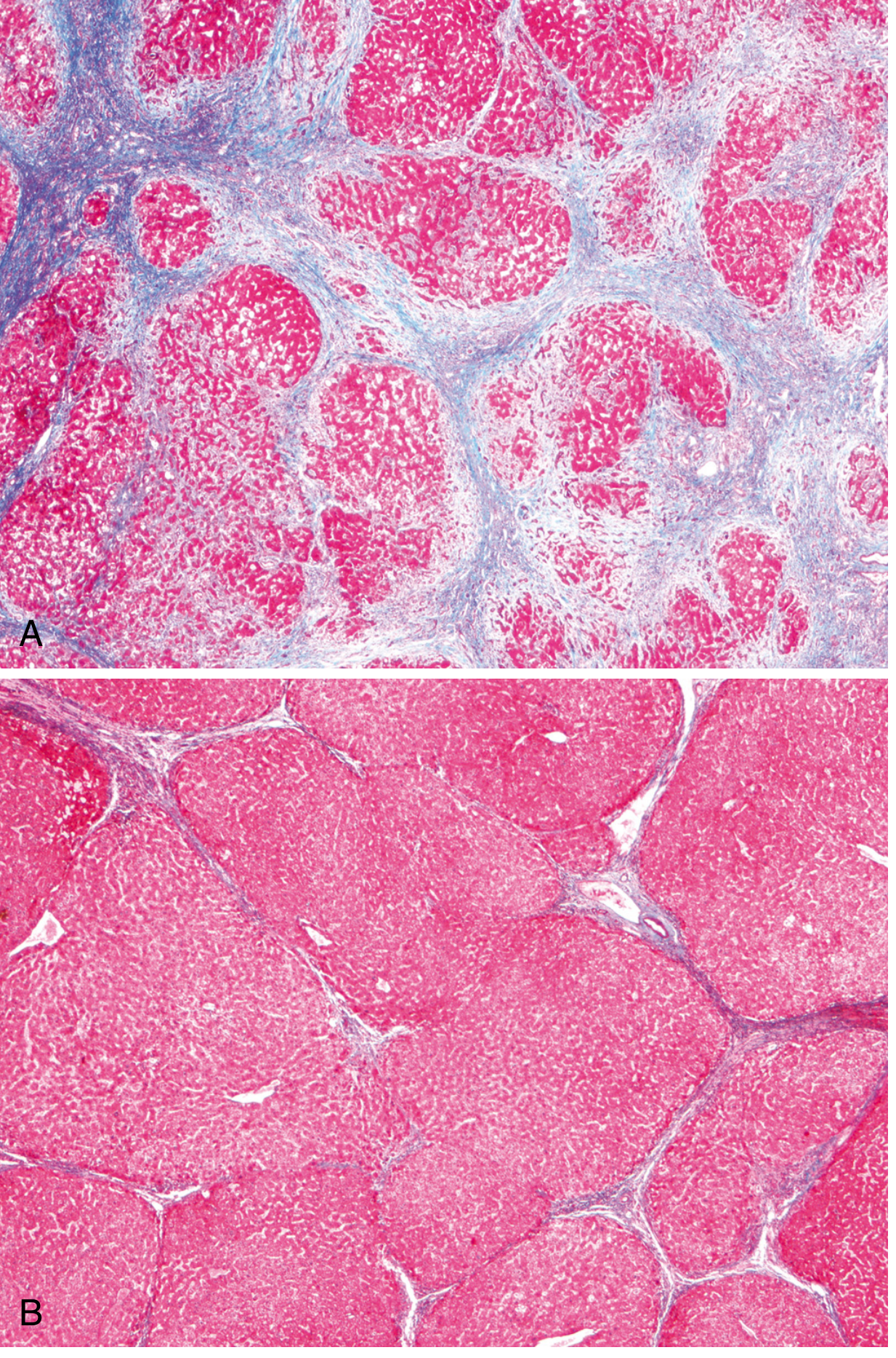
- (A) Active cirrhosis: Thick bands of blue-stained collagen separating rounded cirrhotic nodules
- (B) After 1 year of abstinence: Most scars are gone, thin residual septa remain - demonstrates reversibility
Macronodular cirrhosis with bridging fibrosis (H&E):
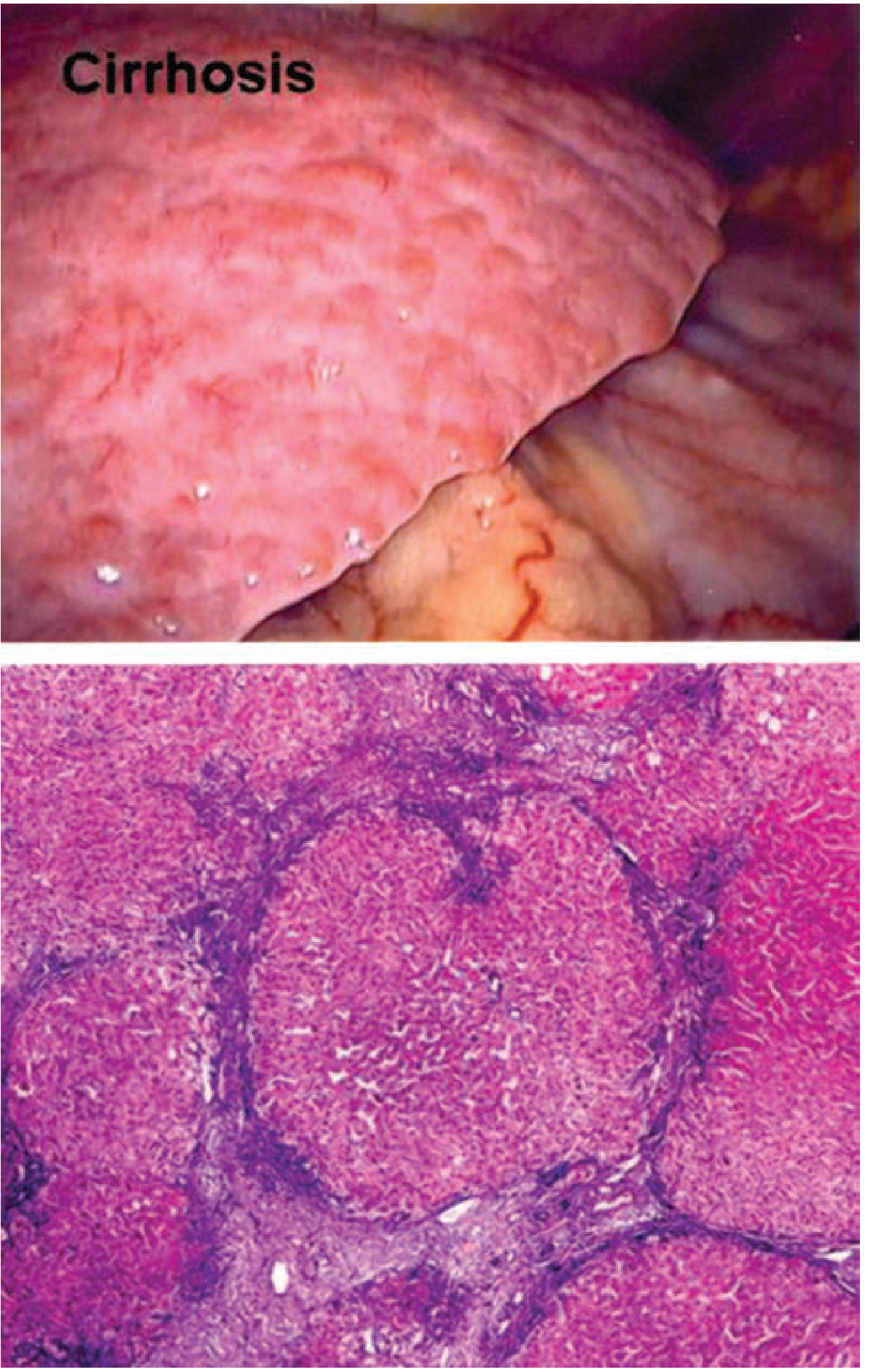
Morphologic Classification (Nodule Size)
| Type | Nodule Size | Pattern | Typical Association |
|---|---|---|---|
| Micronodular | < 3 mm | Thick regular septa, uniform small nodules, all lobules involved | Alcohol-associated (classic), early cirrhosis |
| Macronodular | > 3 mm | Varying nodule and septum size, irregular | Viral hepatitis, PBC, post-necrotic |
| Mixed | Both | Transition pattern | Micronodular converting to macronodular |
Important: This WHO morphologic classification has limited utility - the same pattern can arise from different etiologies, and the same etiology can show multiple patterns. Cirrhosis is a dynamic process in which nodule size changes over time. - Schwartz's Principles of Surgery
5. Consequences and Complications (Pathophysiology)
A. Portal Hypertension
This is the single most important complication and is intrahepatic in origin due to two mechanisms:
- Mechanical obstruction - fibrous bands and regenerative nodules compress and distort sinusoids and portal venules, increasing resistance to portal blood flow
- Increased portal blood flow - peripheral and splanchnic vasodilation (mediated by nitric oxide and other vasodilators) causes a hyperdynamic circulation that further increases portal inflow
Causes of portal hypertension (by location):
| Location | Examples |
|---|---|
| Prehepatic | Portal vein thrombosis, splenomegaly with increased splenic vein flow |
| Intrahepatic (dominant) | Cirrhosis (any cause), nodular regenerative hyperplasia, schistosomiasis, massive fatty change, sarcoidosis |
| Posthepatic | Right heart failure, constrictive pericarditis, hepatic vein outflow obstruction (Budd-Chiari) |
Consequences of portal hypertension:
- Esophageal and gastric varices - collateral vessels at the gastroesophageal junction that can rupture catastrophically. The most feared acute complication - variceal hemorrhage is a leading cause of death.
- Caput medusae - dilated periumbilical collaterals from recanalization of the umbilical vein
- Hemorrhoids - collaterals at the anorectal junction
- Congestive splenomegaly → hypersplenism (thrombocytopenia, anemia, leukopenia)
- Ascites (see below)
B. Ascites
Mechanism (multifactorial):
- Portal hypertension → increased hydrostatic pressure in splanchnic capillaries
- Hypoalbuminemia (liver failure) → reduced oncotic pressure
- Splanchnic vasodilation → arterial underfilling → activation of renin-angiotensin-aldosterone system (RAAS) and ADH → sodium and water retention
C. Hepatic Encephalopathy
- The liver normally clears ammonia (from gut bacteria and amino acid metabolism) by converting it to urea. In cirrhosis + portosystemic shunting, ammonia bypasses the liver and reaches the brain.
- Other neurotoxins (mercaptans, short-chain fatty acids, false neurotransmitters) also contribute.
- Clinically: ranges from subtle personality changes → asterixis (flapping tremor) → coma.
D. Hepatorenal Syndrome
- Renal failure WITHOUT intrinsic renal pathology
- Mechanism: Hepatic failure → excess vasodilator production (nitric oxide) → reduced renal perfusion pressure → activation of renal sympathetic nervous system and RAAS → afferent arteriolar vasoconstriction → progressive renal failure
- Reversible if liver function is restored (e.g., by transplantation)
E. Hyperestrogenemia
Due to impaired hepatic estrogen metabolism:
- Spider angiomas (central pulsating arteriole with radiating vessels)
- Palmar erythema (local vasodilation)
- Gynecomastia and hypogonadism in males
- Menstrual irregularities in females
F. Coagulopathy
- Reduced synthesis of clotting factors (all factors except vWF are made in the liver)
- Thrombocytopenia from hypersplenism
- Vitamin K malabsorption (due to cholestasis)
G. Hepatocellular Carcinoma (HCC)
- Cirrhosis is the strongest risk factor for HCC, regardless of etiology
- Repeated cycles of cell death and regeneration → accumulation of genetic mutations → malignant transformation
- HBV-related HCC can arise without cirrhosis (HBV DNA integrates into the genome)
- Annual ultrasound surveillance is standard of care in cirrhotic patients
H. Other
- Bacterial infections/Spontaneous Bacterial Peritonitis (SBP) - Kupffer cell dysfunction + gut mucosal barrier damage → bacteremia → seeding of ascitic fluid
- Cholestasis and pruritus - persistent pruritus from bile salt accumulation can be profound and debilitating
- Jaundice - impaired bilirubin conjugation and excretion
6. Compensated vs. Decompensated Cirrhosis
| Feature | Compensated | Decompensated |
|---|---|---|
| Definition | Cirrhosis without major complications | Development of ascites, variceal hemorrhage, encephalopathy, or jaundice |
| Median survival | >12 years | ~2 years without transplant |
| Mechanism | Adequate functional reserve | Portal hypertension + liver insufficiency |
Decompensation is defined by any of four events: ascites, variceal hemorrhage, encephalopathy, jaundice - Goldman-Cecil Medicine
7. Clinical Features Summary
About 40% of patients with cirrhosis are asymptomatic until advanced stages. The common causes of death are:
- Hepatic encephalopathy
- Bleeding from esophageal varices
- Bacterial infections (SBP, sepsis)
- Hepatocellular carcinoma
8. Key Pathology Exam Points
| Fact | Detail |
|---|---|
| Hallmark stain | Masson Trichrome (collagen = blue) |
| Central cell in pathogenesis | Hepatic stellate cell (Ito cell) in Space of Disse |
| Transformation | Quiescent stellate cell → contractile myofibroblast |
| Key cytokine | TGF-β1 (pro-fibrotic); PDGF (proliferation) |
| Collagen types deposited | Types I and III |
| Alcohol-associated cirrhosis histology | Micronodular pattern, Mallory bodies (hyaline), neutrophil infiltrate, steatosis |
| Key special stain to highlight nodules | Trichrome or reticulin |
| Key reversibility concept | Fibrosis - even cirrhosis - can regress with removal of the causative agent |
| HCC risk | Present in all forms of cirrhosis; screen with AFP + ultrasound every 6 months |
Sources: Robbins & Cotran Pathologic Basis of Disease (10e), pp. 769-770; Robbins & Kumar Basic Pathology, pp. 597-598; Goldman-Cecil Medicine, p. 1614; Schwartz's Principles of Surgery (11e), p. 1390
This is a shared conversation. Sign in to Orris to start your own chat.