Pneumonitis
Reading File
Finding Sources
Searching PubMed
pneumonitis
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive, well-sourced material. Let me compile the complete answer.
Pneumonitis
Pneumonitis refers to inflammation of lung parenchyma (primarily the alveolar walls and interstitium) caused by a non-infectious trigger. This distinguishes it from pneumonia, where infection is the primary driver. Pneumonitis is a broad umbrella that encompasses several clinically important entities.
Types and Causes
1. Hypersensitivity Pneumonitis (HP)
The most extensively studied form. Also called extrinsic allergic alveolitis.
HP is a complex inflammatory and/or fibrotic interstitial lung disease (ILD) in susceptible individuals, caused by an immunologically mediated reaction to one or more inhaled antigens from occupational, avocational, residential, or environmental settings. - Murray & Nadel's Textbook of Respiratory Medicine
Common antigen sources:
| Source | Exposure | Classic Syndrome |
|---|---|---|
| Thermophilic actinomycetes | Moldy hay, dairy barns | Farmer's lung |
| Bird proteins (feathers, excreta) | Pigeons, parakeets, doves | Pigeon breeder's / Bird fancier's lung |
| Fungi/yeasts | Hot tubs, humidifiers, moldy wood | Hot-tub lung |
| M. avium complex | Metalworking fluids, sauna | Metalworker's lung |
| Isocyanates, zinc, dyes | Industrial chemicals | Chemical HP |
- Robbins Basic Pathology (Robbins Pathology), p. 463
2. Drug-Induced Pneumonitis
Many drugs can cause lung inflammation through direct toxic effects or immunological mechanisms.
- Bleomycin - direct cytotoxicity + inflammatory cell recruitment causing pneumonitis and fibrosis
- Amiodarone - associated with pneumonitis and fibrosis
- Methotrexate - can produce lung infiltrates similar to RA-related lung disease
- Checkpoint inhibitors (immunotherapy) - increasingly recognized cause
Key diagnostic challenge: temporal relationship between drug use and symptoms is often not apparent; latency from drug onset to toxicity is highly variable; some drugs (e.g., amiodarone, nitrofurantoin) can produce acute, subacute, or chronic patterns. - Fishman's Pulmonary Diseases and Disorders
3. Radiation Pneumonitis
A well-known complication of thoracic irradiation. Acute radiation pneumonitis occurs 1-6 months after therapy in up to 20% of patients, manifesting with fever, dyspnea (out of proportion to the irradiated lung volume), pleural effusion, and infiltrates in the irradiated field. With corticosteroid therapy, symptoms may resolve, or the disease may progress to chronic radiation pneumonitis with pulmonary fibrosis. - Robbins Basic Pathology, p. 462
4. HIV-Related Pneumonitis
- Lymphocytic interstitial pneumonitis - classically affects HIV-infected infants; may occur in adults at CD4 >350 cells/μL
- Nonspecific interstitial pneumonitis - often asymptomatic; can mimic Pneumocystis pneumonia at CD4 <200 cells/μL
- Goldman-Cecil Medicine
Immunopathogenesis of HP
Multiple immune mechanisms contribute:
- BAL shows increased CD4+ and CD8+ T lymphocytes and elevated proinflammatory chemokines (MIP-1α / CCL3, IL-8)
- Most patients have specific antibodies (precipitins) against the causative antigen
- Complement and immunoglobulins deposited within pulmonary vessel walls
- Nonnecrotizing granulomas in two-thirds of cases, implying Type IV (T cell-mediated) hypersensitivity - Robbins Pathologic Basis of Disease, p. 715
Both Type III (immune complex) and Type IV (delayed hypersensitivity) mechanisms are operative.
Genetic susceptibility: The MUC5B promoter variant (rs35705950) and short telomere length (<10th percentile for age) are associated with fibrotic HP and reduced transplant-free survival. Approximately 10% of chronic HP patients carry protein-altering variants in telomere-related genes. - Murray & Nadel's
Smoking paradox: Active smokers are less likely to develop HP (reduced immune response), but those who already have HP and continue smoking have a worse prognosis.
Histopathology
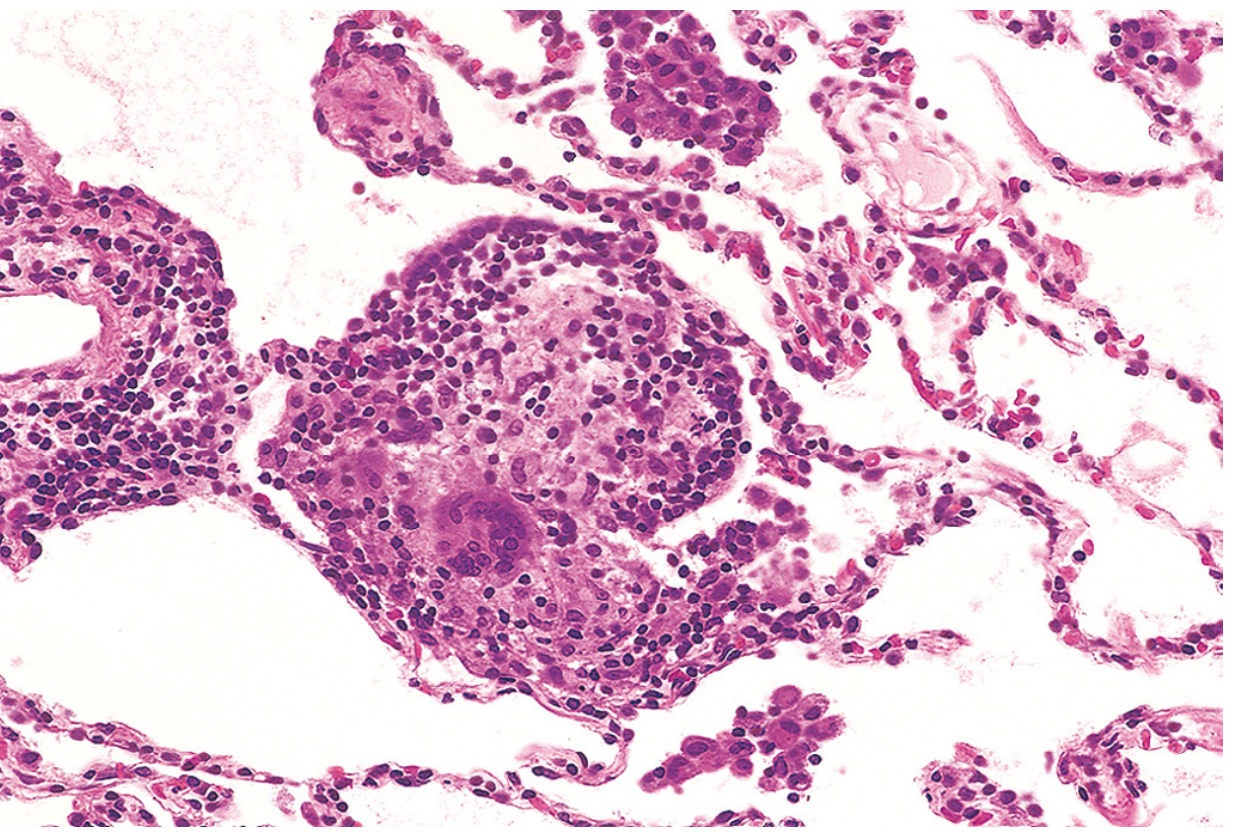
Phase-dependent changes:
- Acute (hours-days): Alveolar damage; patchy interstitial infiltrate of lymphocytes, plasma cells, macrophages; variable neutrophils
- Subacute: Poorly formed peribronchiolar granulomas; interstitial pneumonitis centered on bronchioles; features overlapping NSIP and organizing pneumonia
- Chronic/fibrotic: Interstitial fibrosis, fibroblastic foci, honeycombing, obliterative bronchiolitis - bilateral, often upper-lobe dominant; may show UIP or NSIP pattern
Classification (Current)
HP is now classified into two groups, replacing the old acute/subacute/chronic time-based model:
| Category | Features | Prognosis |
|---|---|---|
| Nonfibrotic (inflammatory) HP | No radiographic fibrosis; ground-glass opacities; reversible | Normalizes with antigen removal ± immunosuppression |
| Fibrotic HP | Reticulation, traction bronchiectasis, honeycombing on HRCT; irreversible | Progressive; does not normalize; higher mortality |
Clinical Features
Nonfibrotic HP:
- Acute flu-like syndrome: malaise, fever, chills arising 4-8 hours after antigen exposure
- Respiratory: nonproductive cough, dyspnea
- Symptoms resolve within 24-48 hours but recur with re-exposure
- Temporal relation to exposure usually recognized
Fibrotic HP:
- Insidious onset: progressive exertional dyspnea, cough, malaise, weight loss
- Temporal relation to exposure often not recognized
- May present as a progressive ILD indistinguishable from IPF
- Velcro-type crackles on auscultation
- Clubbing in some
Diagnosis
Step 1 - Exposure assessment: The single most important step. A thorough occupational, avocational, and residential history is essential. Specific IgG antibodies (serum precipitins) confirm exposure but do not establish causation alone.
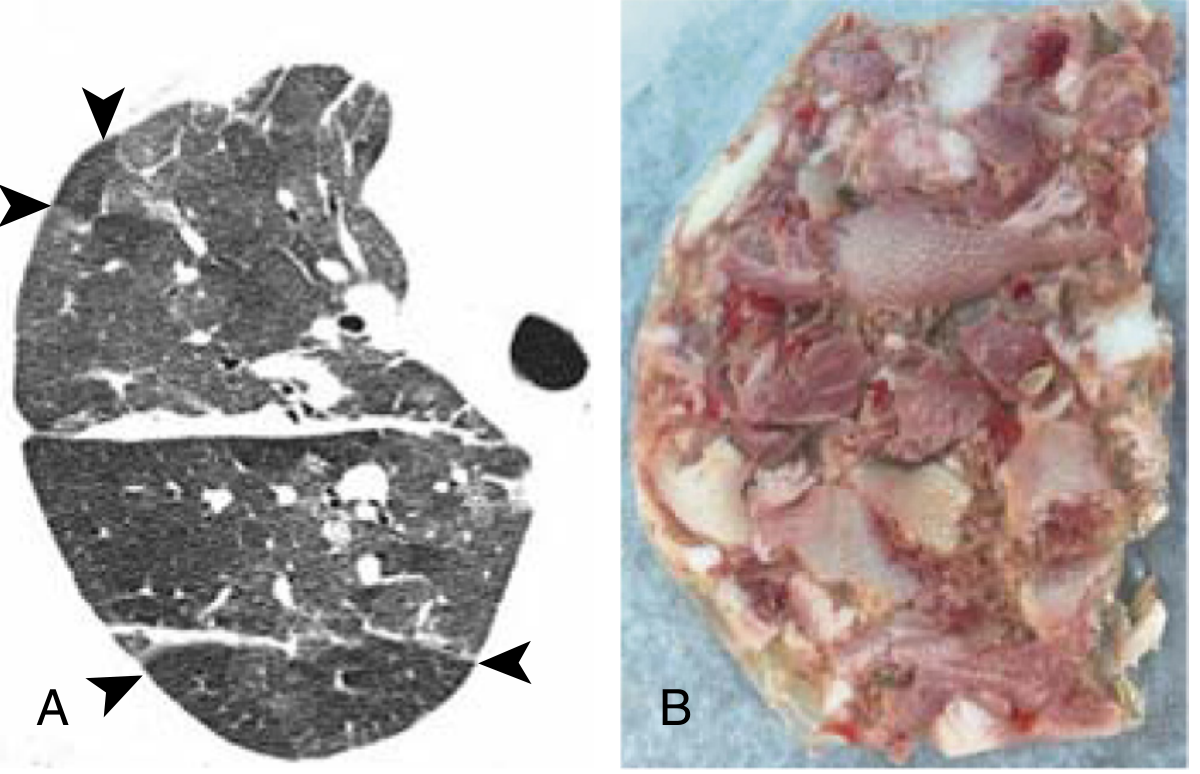
Step 2 - HRCT chest: Required before invasive testing.
- Nonfibrotic: Diffuse bilateral ground-glass opacities, centrilobular nodules, air trapping on expiration
- Fibrotic: Reticulation, traction bronchiectasis, honeycombing, architectural distortion - often upper/mid-lobe predominant
- "Headcheese" sign: Multifocal ground-glass opacity with areas of mosaic perfusion (lobular low attenuation) on the same inspiratory scan, reflecting simultaneous infiltrative and obstructive abnormalities - moderately sensitive (49%) and highly specific (93%) for fibrotic HP
Step 3 - Bronchoalveolar lavage (BAL):
- Lymphocytosis (≥25% lymphocytes) on BAL differential - most common cellular profile in HP and drug-induced ILD
- CD8+ (T suppressor) lymphocyte predominance is common in drug-induced HP; CD4+ predominance in other HP types
- Also used to exclude infection
Step 4 - Lung biopsy (if needed): Transbronchial or surgical lung biopsy for working diagnoses where clinical + HRCT + BAL are insufficient
Gold standard: Multidisciplinary discussion (MDD) integrating clinical, radiologic, and histopathologic findings - as with all ILD.
Pulmonary Function Tests
Primarily restrictive pattern:
- Decreased FVC, TLC, lung compliance
- Decreased DLCO (diffusing capacity for CO)
- In fibrotic HP with coexisting emphysema: DLCO markedly low but lung volumes may be relatively preserved
Prognostic Indicators (Baseline)
Worse prognosis associated with:
- Male sex, older age
- Auscultatory crackles
- Low FVC%, low DLCO%
- Honeycombing, traction bronchiectasis, increased fibrosis on HRCT
- Fibroblast foci on biopsy
- MUC5B variant; short telomere length
- Unknown inciting antigen
- Smoking; acute exacerbation; pulmonary hypertension
Management
1. Antigen Avoidance (cornerstone)
- Identify and remove (or remove patient from) the antigen source
- Nonfibrotic HP: complete resolution expected if antigen is eliminated promptly
- Fibrotic HP: antigen removal alone does not reverse established fibrosis but may slow progression
2. Pharmacologic Therapy
Initiated when clinically significant disease progression occurs despite antigen remediation:
- Corticosteroids (prednisone): First-line; accelerate symptom resolution in acute/subacute forms. Tapering regimen required
- Steroid-sparing agents: Azathioprine (AZA), mycophenolate mofetil (MMF) for chronic fibrotic disease or if steroids cannot be tapered
3. Supportive Measures
- Supplemental oxygen for hypoxemia
- Pulmonary rehabilitation
- Smoking cessation
- Vaccinations (influenza, pneumococcal)
- Management of comorbidities (pulmonary hypertension)
4. Lung Transplantation
- For end-stage fibrotic HP refractory to medical management
- Referral for evaluation if indicated
Drug-Induced Pneumonitis: Diagnostic Approach
- Establish that a recognized drug with known pulmonary toxicity is being used
- Clinical history, exam, labs, and HRCT compatible with known patterns of lung injury
- Exclude infection via BAL culture
- Multidisciplinary approach: pulmonology, radiology, rheumatology, pathology
- Rechallenge is generally not advised; improvement after drug withdrawal supports the diagnosis
Key Differentials
- Infectious pneumonia (bacterial, viral, fungal, Pneumocystis)
- Idiopathic interstitial pneumonias (IPF, NSIP, COP)
- Sarcoidosis
- Eosinophilic pneumonia
- Connective tissue disease-associated ILD
- Drug-induced ILD
Sources:
- Murray & Nadel's Textbook of Respiratory Medicine (Ch. 91 - Hypersensitivity Pneumonitis)
- Robbins & Kumar Pathologic Basis of Disease (Ch. 15)
- Robbins Basic Pathology (Ch. 11)
- Fishman's Pulmonary Diseases and Disorders (Drug-Induced Respiratory Disease)
- Goldman-Cecil Medicine (HIV-related pneumonitis)
This is a shared conversation. Sign in to Orris to start your own chat.