Cell injury
Reading File
Finding Sources
Searching PubMed
"cell injury" mechanisms review
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have everything I need for a comprehensive, well-illustrated answer on cell injury. Let me compile it.
Cell Injury
Robbins & Kumar Basic Pathology (Robbins Pathology)
Overview
Cells constantly adjust their structure and function to accommodate changing demands - a process called homeostasis. When a physiologic stress or noxious stimulus exceeds the cell's adaptive capability, cell injury results. Injury may be:
- Reversible - homeostasis is restored once the stress is removed
- Irreversible - leading to cell death (necrosis or apoptosis)
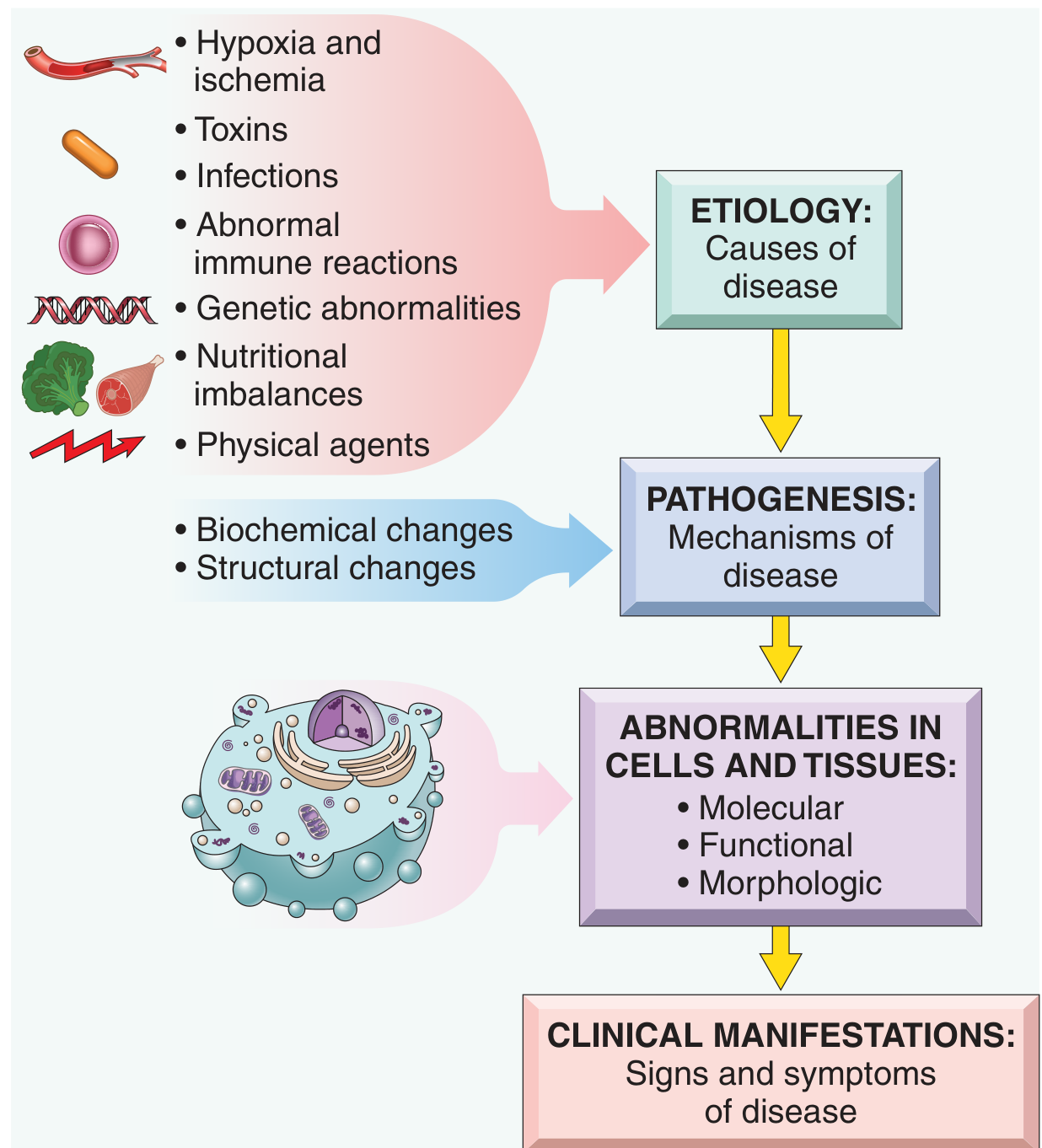
1. Causes of Cell Injury
| Category | Examples |
|---|---|
| Hypoxia and ischemia | Arterial blockage, anemia, pulmonary disease - most common cause |
| Toxins | Air pollutants, CO, asbestos, ethanol, drugs, insecticides |
| Infectious agents | Viruses, bacteria, fungi, parasites - via toxins or immune responses |
| Immunologic reactions | Autoimmunity, allergy, chronic inflammation |
| Genetic abnormalities | Chromosomal defects, enzyme deficiency (inborn errors), sickle cell mutation |
| Nutritional imbalances | Protein-calorie deficiency, vitamin deficiencies, obesity |
| Physical agents | Trauma, heat, cold, radiation, electric shock |
| Aging | Diminished replicative capacity and accumulated damage |
2. Cellular Responses: The Spectrum
The response to injury depends on the type, duration, and severity of the stimulus, as well as the cell type and its metabolic state:
- Adaptation (new steady state preserved): hypertrophy, hyperplasia, atrophy, metaplasia
- Reversible injury: cell swelling, fatty change - cell recovers if stress removed
- Irreversible injury/cell death: necrosis or apoptosis
3. Reversible Cell Injury - Morphology
The earliest ultrastructural changes include:
- Cell swelling (cytoplasmic vacuolation / "hydropic change") - from failure of Na+/K+-ATPase
- Dilation of the endoplasmic reticulum (ER)
- Detachment of ribosomes from rough ER
- Fatty change (lipid vacuoles) - especially in liver, heart, kidney cells dependent on fat metabolism
These are fully reversible if the injurious stimulus is removed.
4. Irreversible Injury and Cell Death
Necrosis
- Characterized by cell swelling, membrane rupture, and release of cellular contents, triggering inflammation
- Morphologically: eosinophilic cytoplasm, nuclear changes (pyknosis, karyorrhexis, karyolysis)
Morphologic patterns of necrosis:
| Type | Characteristics | Common Cause/Location |
|---|---|---|
| Coagulative | Preserved cell outlines, "ghost" cells; protein denaturation | Infarcts of heart, kidney, liver |
| Liquefactive | Digestion of tissue; pus formation | Brain infarcts, bacterial abscesses |
| Caseous | "Cheese-like" appearance; amorphous granular debris | TB granulomas |
| Fat necrosis | Calcium soap deposits (saponification) | Pancreatic/breast fat |
| Gangrenous | Coagulative + liquefactive (dry vs wet) | Limb ischemia |
| Fibrinoid | Bright pink deposits in vessel walls | Immune vasculitis, malignant hypertension |
Apoptosis
- Programmed cell death - cell shrinks, chromatin condenses, cell fragments into apoptotic bodies
- No inflammation (bodies are phagocytosed cleanly)
- Mediated by caspases (executioner proteases)
- Two main pathways:
- Intrinsic (mitochondrial): triggered by DNA damage, ER stress, growth factor withdrawal - regulated by BCL-2 family proteins (pro-apoptotic: BAX, BAK; anti-apoptotic: BCL-2, BCL-XL); releases cytochrome c → activates caspase-9 → caspase-3
- Extrinsic (death receptor): Fas-L binds Fas (CD95) or TNF binds TNFR → FADD → caspase-8 → caspase-3
5. Mechanisms of Cell Injury
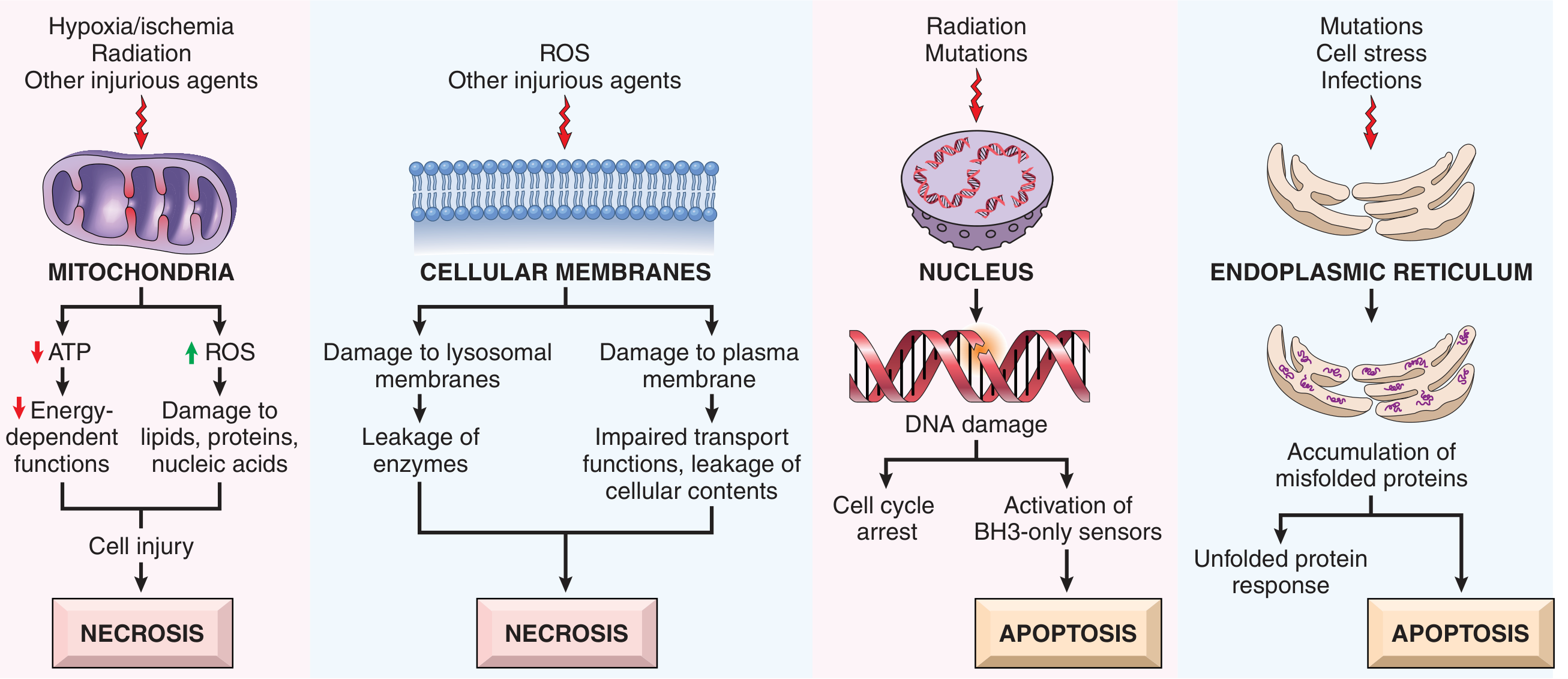
A. Mitochondrial Dysfunction (→ Necrosis)
- Hypoxia, toxins, radiation damage mitochondria → ↓ ATP
- Consequences of ATP depletion:
- Failure of Na+/K+-ATPase → Na+ and water influx → cell swelling
- Anaerobic glycolysis → lactic acid accumulation → ↓ intracellular pH → enzyme inhibition
- Detachment of ribosomes from rough ER → ↓ protein synthesis
- Ultimately: membrane rupture → necrosis
- Mitochondria also generate a permeability transition pore (mPTP) under injury, releasing cytochrome c (triggers apoptosis)
B. Oxidative Stress / Reactive Oxygen Species (ROS)
- ROS sources: mitochondrial leakage, activated phagocytes (respiratory burst), reperfusion after ischemia, radiation
- ROS species: superoxide (O₂⁻), hydrogen peroxide (H₂O₂), hydroxyl radical (·OH)
- Damage caused: lipid peroxidation of membranes, protein oxidation/cross-linking, DNA strand breaks
- Cellular defenses: superoxide dismutase (SOD), catalase, glutathione peroxidase, vitamins E/C
C. Membrane Damage (→ Necrosis)
- Plasma membrane damage: loss of osmotic balance, leakage of cellular contents (enzymes detected in serum: troponin, CK-MB, LDH, AST)
- Lysosomal membrane damage: release of digestive enzymes → autodigestion of cell (endonucleases, proteases, phospholipases)
- Mechanisms: direct toxins, ROS-mediated lipid peroxidation, decreased phospholipid synthesis, loss of membrane phospholipids
D. Calcium Homeostasis Disturbance
- Normally, intracellular Ca²+ is very low (0.1 μM) vs. extracellular (1.3 mM)
- Ischemia and toxins → cytosolic Ca²+ rises → activates:
- Phospholipases (membrane damage)
- Proteases (cytoskeletal/membrane breakdown)
- ATPases (↓ ATP)
- Endonucleases (DNA fragmentation)
E. Endoplasmic Reticulum (ER) Stress (→ Apoptosis)
- Accumulation of misfolded proteins → unfolded protein response (UPR)
- If UPR cannot restore normal ER function → activates apoptosis
- Relevant in: diabetes (β-cell loss), neurodegeneration (Alzheimer's, Parkinson's), atherosclerosis
F. DNA Damage (→ Apoptosis)
- Caused by radiation, ROS, chemotherapy drugs
- Activates p53 → either cell cycle arrest (to allow DNA repair) or apoptosis if damage is irreparable
- Activates BH3-only sensors → triggers intrinsic apoptosis pathway
6. Clinicopathologic Examples
Hypoxia/Ischemia
- Sequence: ↓O₂ → ↓ATP → cell swelling (reversible) → membrane failure → necrosis (irreversible)
- Point of no return: mitochondrial membrane permeability transition; lysosomal rupture
Ischemia-Reperfusion Injury
- Paradoxically, restoring blood flow can worsen injury
- Mechanisms: burst of ROS on reoxygenation, Ca²+ overload, inflammatory cell recruitment, complement activation
- Clinically important in: MI reperfusion therapy, stroke thrombolysis, organ transplantation
Toxic Cell Injury
- Direct toxicity: e.g., HgCl₂ binds sulfhydryl groups on membrane proteins → membrane injury
- Bioactivation: e.g., CCl₄ → CCl₃ radical (by P-450) → lipid peroxidation → fatty liver → necrosis
- Acetaminophen overdose: hepatotoxic NAPQI metabolite accumulates when glucuronidation is saturated
7. Autophagy
- A survival mechanism under nutrient deprivation - cellular components are sequestered in autophagosomes, fused with lysosomes, and digested to provide nutrients
- Can paradoxically trigger apoptosis if excessive
- Regulated by ATG (autophagy) genes
Summary Table: Reversible vs. Irreversible Injury
| Feature | Reversible | Irreversible |
|---|---|---|
| Cell swelling | + | ++ |
| Membrane damage | Plasma membrane bleb | Rupture |
| Mitochondria | Swollen, small densities | Large densities, rupture |
| Nuclear changes | None/mild | Pyknosis, karyorrhexis, karyolysis |
| Ribosomes | Detach from RER | Disaggregate |
| ATP | ↓ | ↓↓↓ |
| Outcome | Recovery | Necrosis or apoptosis |
Source: Robbins & Kumar Basic Pathology (Robbins Pathology), pp. 16-129
This is a shared conversation. Sign in to Orris to start your own chat.