Pathogenesis of breast cancer
Reading File
Finding Sources
Finding Sources
Searching PubMed
"breast cancer"[MeSH Terms] AND "pathogenesis"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive content. Let me compile the complete answer.
Pathogenesis of Breast Cancer
Based on Robbins, Cotran & Kumar - Pathologic Basis of Disease
Overview
Breast cancers arise through the stepwise acquisition of driver mutations in epithelial cells. The initiating mutation strongly influences the phenotype of the cancer that ultimately develops. Two largely separate evolutionary pathways - low-grade and high-grade - diverge early and produce distinct tumor subtypes with different molecular profiles, histologic appearances, and clinical behaviors.
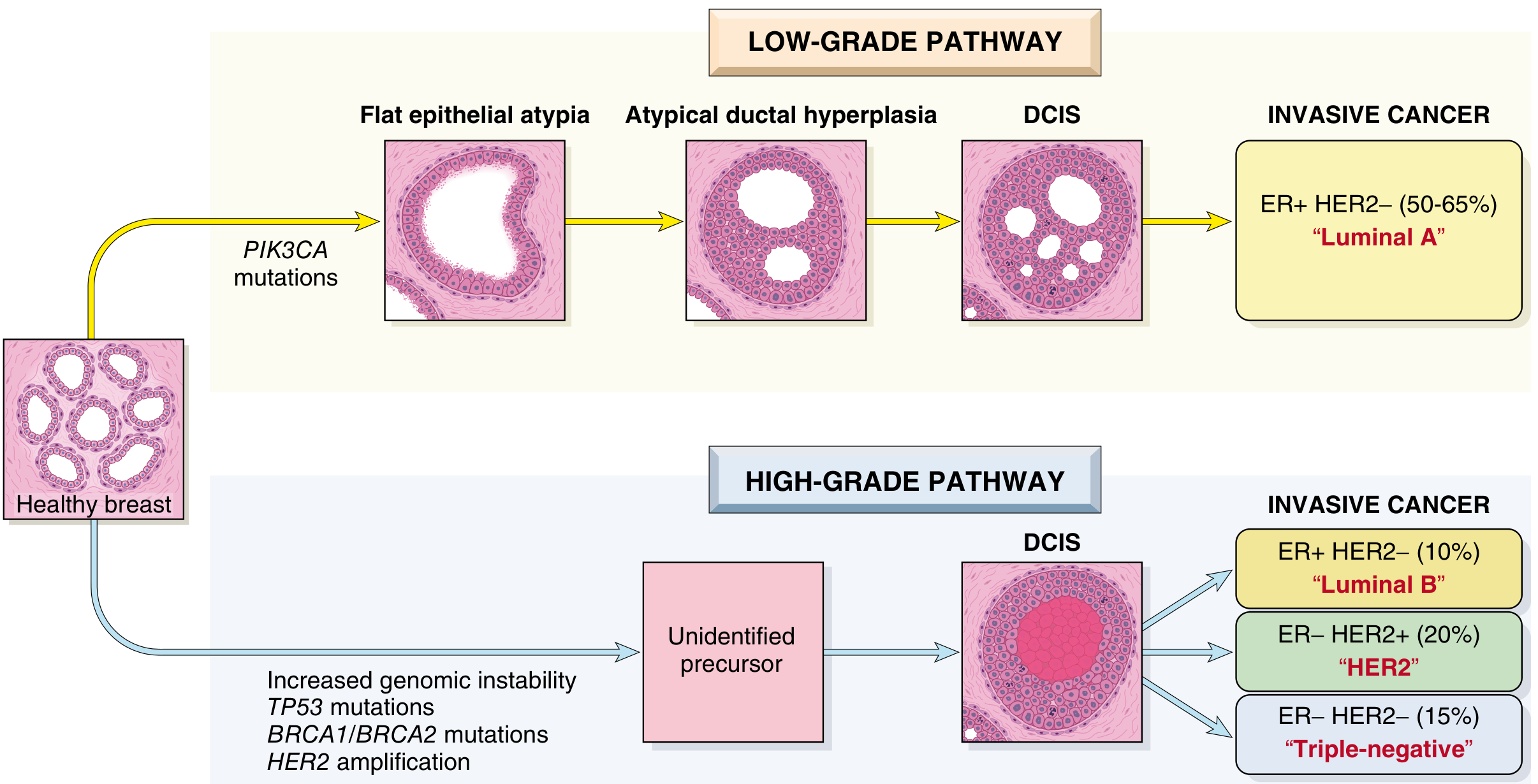
Fig. 23.17 - The two major pathways of breast cancer evolution stratified by histological grade
1. The Two Major Evolutionary Pathways
Low-Grade Breast Neoplasia Pathway
This pathway proceeds through recognizable ER-positive precursor lesions in a stepwise fashion:
Columnar cell lesions → Flat epithelial atypia → Atypical hyperplasia (ductal/lobular) → Low-grade DCIS / classic LCIS → Invasive ER+ HER2- cancer ("Luminal A", 50-65%)
Key molecular features:
- Loss of 16q and gain of 1q are the defining genomic alterations
- PIK3CA mutations are common early events
- Predominantly "luminal" gene expression signature
- Low genomic instability
High-Grade Breast Neoplasia Pathway
The major precursor lesion is high-grade DCIS; the earliest steps are not fully characterized.
Unidentified precursor → High-grade DCIS → Three divergent invasive subtypes:
- ER+ HER2- "Luminal B" (~10%)
- ER- HER2+ "HER2 subtype" (~20%)
- ER- HER2- "Triple-negative" (~15%)
Key molecular features:
- TP53 mutations (very common)
- Loss of 13q, gain of 11q13, amplification of 17q12
- BRCA1/BRCA2 loss (especially in TNBC)
- HER2 amplification (in HER2+ subtype)
- Gene expression enriched for proliferation-associated genes
- High genomic instability
2. Transition from In Situ to Invasive Carcinoma
The molecular drivers of this transition remain poorly understood. Most genomic changes seen in invasive carcinoma are already present in in situ precursors, including major driver mutations - no changes specific to invasion have been identified. This suggests that stromal/microenvironmental changes, rather than further tumor cell mutations, may drive invasion. - Robbins, Cotran & Kumar, p. 972
3. The Tumor Microenvironment
The tumor microenvironment plays an important role in breast cancer initiation, progression, and dissemination:
- Myoepithelial cells: normally maintain the basement membrane; their reduction in number in some in situ cancers creates gaps that may set the stage for stromal invasion
- Cancer-associated fibroblasts (CAFs): promote tumor progression by facilitating cell adhesion, ECM remodeling, and immunosuppression
- Immune checkpoint upregulation: breast cancer cells upregulate PD-L1 and other surface ligands to suppress the host immune response
- Together, these interactions create a "tumor-promoting microenvironment" that allows cancer cells to evade immunity
4. Pathogenesis of Familial Breast Cancer
~25%-35% of breast cancers arise in individuals with a susceptibility gene variant. Single gene mutations with moderate-to-high penetrance account for 8%-17% of all breast carcinomas.
BRCA1 and BRCA2 (Most Important)
- Responsible for 80%-90% of single-gene familial breast cancers; ~3%-6% of all breast cancers
- BRCA1 (chr 17q21): ~55% of hereditary single-gene cases; risk of 40%-90% by age 70; majority of tumors are triple-negative
- BRCA2 (chr 13q12.3): ~35% of hereditary single-gene cases; risk of 30%-60%; majority are ER-positive
- Both genes function in homologous recombination (repair of double-strand DNA breaks)
- Tumors with homologous recombination deficiency are susceptible to PARP inhibitors, which block single-strand repair - loss of both pathways is lethal to tumor cells ("synthetic lethality")
- In Ashkenazi Jewish populations, ~1 in 40 individuals carries one of three founder mutations
Other High-Penetrance Genes (Table 23.3)
| Gene (Syndrome) | Risk by Age 70 | Tumor Type | Notes |
|---|---|---|---|
| PALB2 | 30%-60% | ER+ or TNBC | Partner of BRCA2; Fanconi anemia if biallelic |
| TP53 (Li-Fraumeni) | 50%-60% | ER+, HER2+ | Also sarcomas, brain tumors, leukemia |
| PTEN (Cowden) | 20%-80% | Apocrine differentiation | Also thyroid, endometrium |
| STK11 (Peutz-Jeghers) | 40%-60% | Various | Also GI polyps |
| CDH1 | ~50% | Lobular carcinoma | Also hereditary diffuse gastric cancer |
Moderate-Penetrance Genes
- ATM (ataxia-telangiectasia): 15%-30% risk; ER-positive tumors
- CHEK2: 10%-30% risk; ER-positive tumors; also prostate, colon, kidney
- RAD51C / RAD51D: 17%-30% risk; majority are TNBC
All of the above genes (BRCA1, BRCA2, PALB2, ATM, RAD51C, RAD51D, CHEK2) share a common function - homologous recombination repair - explaining the common thread in hereditary susceptibility.
5. Pathogenesis of Sporadic Breast Cancer
The three major molecular subtypes and their sporadic pathways:
A. Luminal A (ER+/PR+, HER2-, Low Grade) - Most Common
- Driven by estrogen signaling: estrogen receptor activation promotes transcription of proliferation genes (cyclin D1) and survival signals
- Arise via the low-grade pathway
- Key somatic mutations: PIK3CA (PI3K/AKT/mTOR pathway), CDH1 loss (in lobular carcinoma)
- Lobular carcinoma invariably shows E-cadherin (CDH1) loss, explaining the single-file infiltrative growth pattern
B. HER2-Enriched (ER-, HER2+, High Grade)
- Driven by HER2 (ERBB2) amplification at 17q12 (~20% of all breast cancers)
- HER2 is a receptor tyrosine kinase; its amplification activates the PI3K/AKT and RAS/MAPK proliferation cascades
- Arise via the high-grade pathway; high genomic instability and frequent TP53 mutations
- HER2 amplification is both a driver and a therapeutic target (trastuzumab, pertuzumab, lapatinib)
C. Triple-Negative Breast Cancer / Basal-Like (ER-, PR-, HER2-)
- Most aggressive subtype; arise via the high-grade pathway
- Characterized by BRCA1 inactivation (either germline or somatic), TP53 mutations, high genomic instability
- Express basal markers (cytokeratin 5/6, EGFR) - reflecting a basaloid cell of origin
- No targeted therapies except immunotherapy (PD-L1 inhibitors) and PARP inhibitors (for BRCA1/2-mutant cases)
6. Key Molecular Pathways Summary
| Pathway | Role | Targetable |
|---|---|---|
| Estrogen receptor (ER) signaling | Proliferation in luminal cancers | Yes (tamoxifen, aromatase inhibitors) |
| PI3K/AKT/mTOR | Survival, proliferation (PIK3CA mutations common) | Yes (everolimus, alpelisib) |
| HER2/ERBB2 amplification | Uncontrolled RTK signaling | Yes (trastuzumab, T-DM1) |
| BRCA1/2 homologous recombination | DNA repair; loss drives genomic instability | Yes (PARP inhibitors) |
| TP53 loss | Loss of cell cycle checkpoint; drives high-grade tumors | No direct target |
| CDH1 (E-cadherin) loss | Lobular morphology, single-file invasion | No |
| PD-L1 / immune checkpoints | Immune evasion | Yes (pembrolizumab) |
Source: Robbins, Cotran & Kumar - Pathologic Basis of Disease, Chapter 23 (Breast), pp. 971-974
This is a shared conversation. Sign in to Orris to start your own chat.