how does hemochromatosis affect liver in detailed information
Reading File
Finding Sources
Finding Sources
Searching PubMed
"hemochromatosis"[MeSH Terms] AND "liver"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Reading File
Reading File
Loading Image
Finding Sources
Loading Image
Excellent - I now have comprehensive content and key images. Let me compile the full, detailed answer.
Hemochromatosis and the Liver: Detailed Overview
Hemochromatosis is the most important storage disorder affecting the liver. The liver is the primary target organ because it is the main site of iron storage - about 98% of hepatic iron is normally held within hepatocytes. In severe hemochromatosis, total body iron can exceed 50 g, with more than one-third accumulating in the liver (compared to the normal ~0.5 g of hepatic iron storage).
1. Pathogenesis: How Iron Injures the Liver
The fundamental defect is loss of the hepcidin-ferroportin negative feedback loop that normally limits iron absorption:
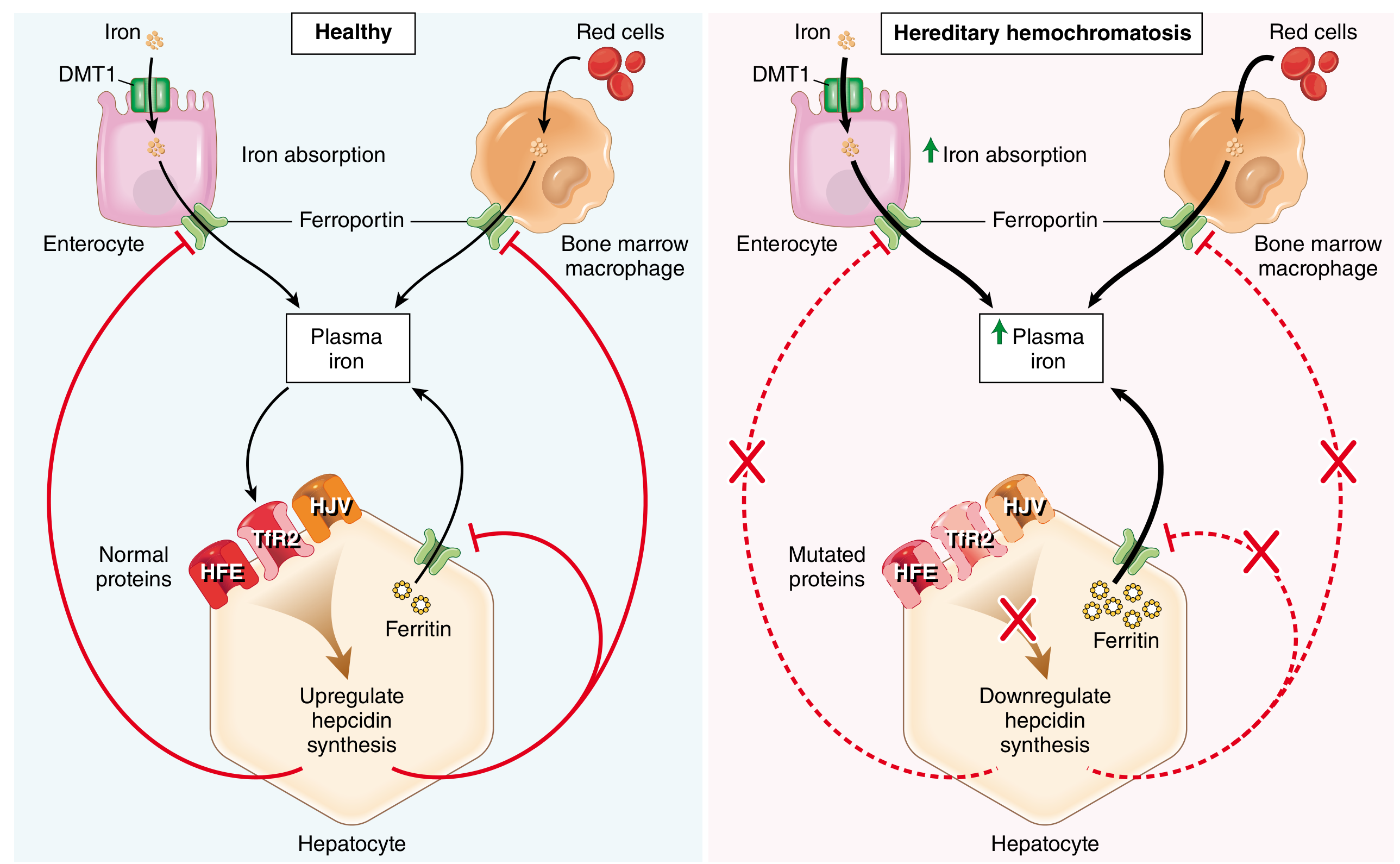
- In normal physiology, hepatocytes sense circulating iron via a sensor complex of HFE, hemojuvelin (HJV), and transferrin receptor 2 (TfR2). When iron is plentiful, this complex stimulates production of hepcidin (encoded by HAMP). Hepcidin binds ferroportin on enterocytes and macrophages, causing its internalization and degradation, which limits iron efflux into the bloodstream.
- In hereditary hemochromatosis, loss-of-function mutations in HFE, HJV, TFR2, or HAMP cripple hepcidin synthesis. Ferroportin activity goes unchecked, and iron pours into the circulation at a rate of 0.5-1 g/year. Disease typically becomes clinically apparent after 20 g of total stored iron has accumulated.
Mechanisms of Hepatocyte Injury (Three Core Pathways)
Once excessive iron accumulates in hepatocytes, it injures cells through three distinct mechanisms:
| Mechanism | Detail |
|---|---|
| Lipid peroxidation | Iron catalyzes free radical (Fenton) reactions that damage hepatocyte cell membranes via lipid peroxidation |
| Stellate cell activation | Iron-generated ROS stimulate hepatic stellate cells, triggering collagen synthesis and progressive fibrosis |
| DNA damage | ROS and iron directly interact with nuclear DNA, causing lethal cell injury and creating a mutagenic environment that predisposes to hepatocellular carcinoma |
Critically, the deleterious effects are reversible if iron is removed early (before cirrhosis). - Robbins & Kumar Basic Pathology, p. 614
2. Genetics of the Most Common Form
The HFE gene (encoding an HLA class I-like molecule) is mutated in >70% of hereditary hemochromatosis cases. The key mutations:
- C282Y (cysteine→tyrosine at position 282): the dominant pathogenic mutation, found almost exclusively in people of European descent. Homozygosity frequency is ~1 in 220 persons of European origin, but only ~10% of homozygous males develop frank iron overload - indicating low penetrance.
- H63D (histidine→aspartate at position 63): worldwide distribution; compound heterozygosity (C282Y/H63D) causes milder disease.
- Juvenile hemochromatosis arises from HAMP or HJV mutations - much rarer, more severe, manifests in young adults.
- Ferroportin disease: SLC40A1 mutations give a different phenotype (macrophage-predominant iron loading).
Because females lose iron through menstrual bleeding, they accumulate iron more slowly, and the male-to-female ratio of clinical disease is 5 to 7:1. - Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 788
3. Hepatic Morphology: What Happens Step by Step
The liver undergoes a characteristic, stage-dependent sequence of changes:
Stage 1 - Early Hemosiderin Deposition
Iron appears first as golden-yellow hemosiderin granules in periportal hepatocytes (zone 1 of the lobule). On Prussian blue stain, these granules appear bright blue:
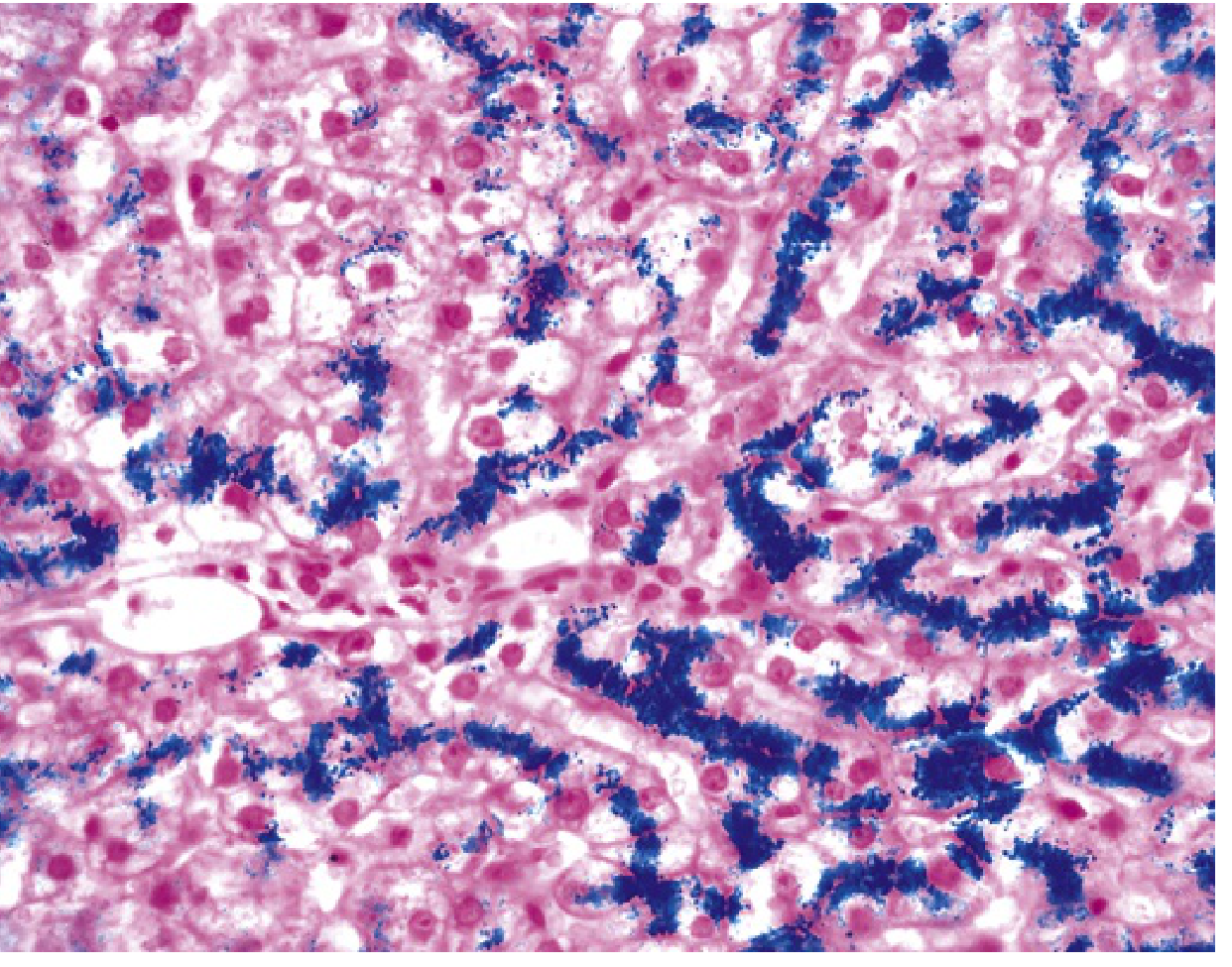
Hereditary hemochromatosis: Prussian blue-stained liver section showing iron deposits (blue) in hepatocytes. At this stage, parenchymal architecture is preserved despite heavy iron loading.
Stage 2 - Progressive Lobular Deposition
With increasing iron burden, deposition spreads from periportal hepatocytes throughout the entire lobule, then into:
- Bile duct epithelium
- Kupffer cells (later than hepatocytes)
The liver becomes slightly enlarged and takes on a characteristic chocolate-brown color from hemosiderin accumulation.
Stage 3 - Fibrosis
Iron-driven stellate cell activation produces fibrous septa that slowly bridge portal tracts to each other (portal-portal bridging fibrosis). This stage is still partly reversible with treatment.
Stage 4 - Cirrhosis
The end-stage is a micronodular cirrhosis in an intensely pigmented (very dark brown to black) liver. The cirrhotic liver from hemochromatosis is grossly distinctive:

Cirrhotic liver from a patient with hemochromatosis. The upper specimen has been stained for iron (blue), illustrating diffuse iron distribution throughout the parenchyma. - Yamada's Textbook of Gastroenterology, p. 2012
Cirrhosis develops in all fully symptomatic patients and is present in 75-80% of clinical hemochromatosis cases.
4. Clinical Liver Features
Hepatomegaly
Hepatomegaly is one of the most common physical signs in hemochromatosis and may be present even before liver enzyme abnormalities. However, it may be absent in young asymptomatic homozygotes.
Liver Enzyme Abnormalities
The liver has a large capacity to accumulate iron before biochemical derangement is seen. Notably, the probability of C282Y hemochromatosis actually decreases as liver enzymes become more abnormal - very elevated transaminases should prompt consideration of coexistent alcoholic liver disease, viral hepatitis, or NASH rather than hemochromatosis alone. Cirrhotic patients and those with concomitant alcohol abuse are more likely to have elevated liver enzymes. - Yamada's Gastroenterology, p. 2011
Cirrhosis and Portal Hypertension
Once established, cirrhosis produces the full spectrum of complications:
- Portal hypertension with esophageal varices
- Ascites and spontaneous bacterial peritonitis
- Hepatic encephalopathy
- Coagulopathy
Liver disease is the most common cause of mortality in HFE-associated hereditary hemochromatosis. - Goldman-Cecil Medicine
5. Hepatocellular Carcinoma (HCC)
The liver in hemochromatosis with cirrhosis carries a dramatically elevated HCC risk:
- HCC occurs in ~18.5% of cirrhotic hemochromatosis patients
- HCC is rare in non-cirrhotic hemochromatosis - cirrhosis is the key prerequisite
- The mutagenic mechanism involves iron-catalyzed ROS causing direct DNA damage and creating pro-carcinogenic conditions
- Coexisting risk factors (alcohol, viral hepatitis) multiply the risk substantially
- Phlebotomy after cirrhosis is established does NOT prevent HCC development (though it reduces the risk when given before cirrhosis occurs)
- Guidelines recommend ultrasound surveillance every 6 months in cirrhotic hemochromatosis patients
Important: Once cirrhosis is present, 30% of patients may develop HCC. - Andrews' Diseases of the Skin, p. Clinical Dermatology
6. Secondary Hemochromatosis and the Liver
Secondary (acquired) hemochromatosis causes identical liver injury patterns. It occurs in:
- Multiple blood transfusions (e.g., beta-thalassemia, sickle cell disease, aplastic anemia)
- Chronic ineffective erythropoiesis: elevated erythroferrone suppresses hepcidin, causing iron overload without genetic mutation
In beta-thalassemia, the combination of transfusional iron loading and erythroferrone-mediated hepcidin suppression makes liver iron accumulation particularly severe.
7. Diagnosis: Liver-Specific Testing
| Test | Finding |
|---|---|
| Serum transferrin saturation | >45% (early, sensitive marker) |
| Serum ferritin | Markedly elevated (reflects iron stores) |
| Liver biopsy | Gold standard - shows Prussian blue-positive deposits, grades fibrosis |
| Hepatic Iron Index (HII) | Iron concentration (µmol/g dry weight) / age; >1.9 suggests hereditary form |
| MRI liver | Non-invasive iron quantification (T2* signal loss) |
| HFE genotyping | Confirms C282Y/H63D mutations; done before biopsy in uncomplicated cases |
Liver biopsy is particularly important when ferritin >1000 µg/L (associated with cirrhosis in ~50% of cases), age >40, or coexistent liver disease is suspected.
8. Treatment and Liver Outcomes
Phlebotomy (First-Line)
- Weekly or twice-weekly removal of 450-500 mL blood (removes ~200-250 mg iron per session)
- Goal: serum ferritin <50 µg/L, transferrin saturation <30%
- Proven benefit: improvement in liver fibrosis, reduced risk of HCC, improved survival in non-cirrhotic disease
- Liver function and hepatomegaly often improve; fibrosis may regress if not yet cirrhotic
- Does NOT reverse established cirrhosis, but prevents progression
Erythrocytapheresis
More efficient iron removal than phlebotomy (red cells extracted, plasma returned), but higher cost.
Chelation
Deferoxamine is NOT recommended for hemochromatosis - inefficient, cumbersome, and potentially toxic. Oral deferasirox has been studied but side effects and cost limit use.
Liver Transplantation
Indicated for decompensated cirrhosis from hemochromatosis. Pre-transplant phlebotomy improves cardiac function. Recurrence of iron overload post-transplant is rare if the native liver's hepcidin deficiency was the driving factor.
Dietary Considerations
- Avoid oral iron supplements and alcohol
- No strict dietary iron restriction (much food iron has poor bioavailability)
- Avoid raw shellfish (risk of Vibrio septicemia in iron overload)
- High-dose vitamin C supplementation is hazardous (mobilizes iron and potentiates cardiac toxicity)
Summary
The liver in hemochromatosis undergoes a predictable, stepwise progression: periportal iron deposition → lobular spread → fibrosis → micronodular cirrhosis → risk of HCC. The key injurious mechanisms are lipid peroxidation, stellate cell-mediated fibrosis, and DNA damage from ROS. Liver disease is the leading cause of death. Early diagnosis (transferrin saturation + HFE genotyping) and timely phlebotomy can completely prevent cirrhosis and normalize life expectancy. Once cirrhosis develops, HCC surveillance is mandatory.
Sources: Robbins & Kumar Basic Pathology; Robbins, Cotran & Kumar Pathologic Basis of Disease; Yamada's Textbook of Gastroenterology (7th ed.); Goldman-Cecil Medicine; Braunwald's Heart Disease (15th ed.)
This is a shared conversation. Sign in to Orris to start your own chat.