Mazabraud syndrome
Reading File
Finding Sources
Searching PubMed
Mazabraud syndrome fibrous dysplasia myxoma
Searching the Web
Mazabraud syndrome clinical features GNAS mutation management 2024
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading Web Page
Mazabraud syndrome pathology imaging treatment prognosis
Loading Image
I now have comprehensive information from multiple authoritative sources. Here is a full synthesis:
Mazabraud Syndrome
Definition
Mazabraud syndrome is a rare condition defined by the dyad of:
- Fibrous dysplasia (FD) of bone - most commonly polyostotic
- Intramuscular myxoma(s) - one or more soft-tissue myxomas in skeletal muscle
It was described by Albright in 1937 and later fully characterized by French surgeon Alain Mazabraud in the 1960s.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 1102
- Campbell's Operative Orthopaedics 15th Ed, p. 1166
Molecular Pathogenesis
The syndrome results from somatic (postzygotic) gain-of-function mutations in the GNAS gene (chromosome 20q13.32), most commonly a missense mutation at codon 201 (R201H or R201C), and rarely codon 227.
- GNAS encodes the alpha subunit of the stimulatory G-protein (Gsα)
- The R201 mutation markedly reduces intrinsic GTPase activity of Gsα
- This causes constitutive activation of Gsα → sustained elevation of cyclic AMP (cAMP)
- Elevated cAMP promotes fibroblast proliferation and inhibits osteoblast differentiation, producing fibrous dysplasia lesions in bone and expansile myxoid masses within muscle
- Fibrous dysplasia lesions also elaborate FGF23, which inhibits renal phosphate resorption, causing hypophosphatemia and hyperphosphaturia
Because the mutation is postzygotic (somatic mosaicism), no inherited case has ever been reported - a germline GNAS mutation is presumed embryonic lethal. The extent and distribution of disease depends on the timing of the mutation during embryogenesis and the tissues affected.
GNAS mutations were confirmed in myxoma tissue in 83% of tested patients in a large European multicenter study (EMSOS multicenter study, PMID: 30653046).
Epidemiology
- Prevalence: ~2.2% among patients with fibrous dysplasia (based on a cohort of 1,446 FD patients from 6 European centers)
- Mean diagnosis of Mazabraud syndrome is ~10 years after the initial FD diagnosis
- Female predominance (22:10 female:male ratio in the EMSOS study)
- Most patients are diagnosed in adulthood (4th-7th decade for myxoma appearance)
- Fibrous dysplasia is often unilateral and more frequently right-sided, consistent with postzygotic mutation
Clinical Features
Fibrous dysplasia component:
- Usually polyostotic (involvement of multiple bones), though monostotic cases occur
- Skeletal features typically manifest in childhood
- Commonly affects: femur, tibia, skull/craniofacial bones, ribs, shoulder and pelvic girdles
- Symptoms: bone pain, pathologic fracture, progressive deformity ("shepherd's crook" deformity of the proximal femur)
- Lesions may continue to enlarge until skeletal maturity
Intramuscular myxoma component:
- Appears in adulthood (often 10+ years after FD diagnosis)
- Usually multiple and located in the same anatomical region as the bone lesions (often the thigh, in proximity to femoral FD)
- The upper thigh/quadriceps muscle is the most frequent site
- Most are asymptomatic or cause mild local pressure/pain
- Rarely cause compressive symptoms or limb deformity
Mazabraud syndrome presents with skeletal features of polyostotic fibrous dysplasia in childhood followed by the appearance of intramuscular myxomas in adulthood, often in the same anatomic region as existing fibrous dysplasia.-- Robbins, Cotran & Kumar, p. 1103
Overlap with McCune-Albright syndrome: 15% of Mazabraud patients also carry features of McCune-Albright syndrome (which adds cafe-au-lait spots and hyperfunctional endocrinopathy to the FD). The three syndromes form a clinical spectrum driven by the same GNAS mutation, differing in timing and tissue distribution.
Imaging
Fibrous dysplasia on plain radiograph/CT:
- Well-defined, homogeneous intramedullary lytic lesion
- Classic "ground-glass" opacification (though not always present)
- Endosteal scalloping, cortical thinning, bone expansion
- No periosteal reaction
- MRI is generally not the recommended primary modality for FD
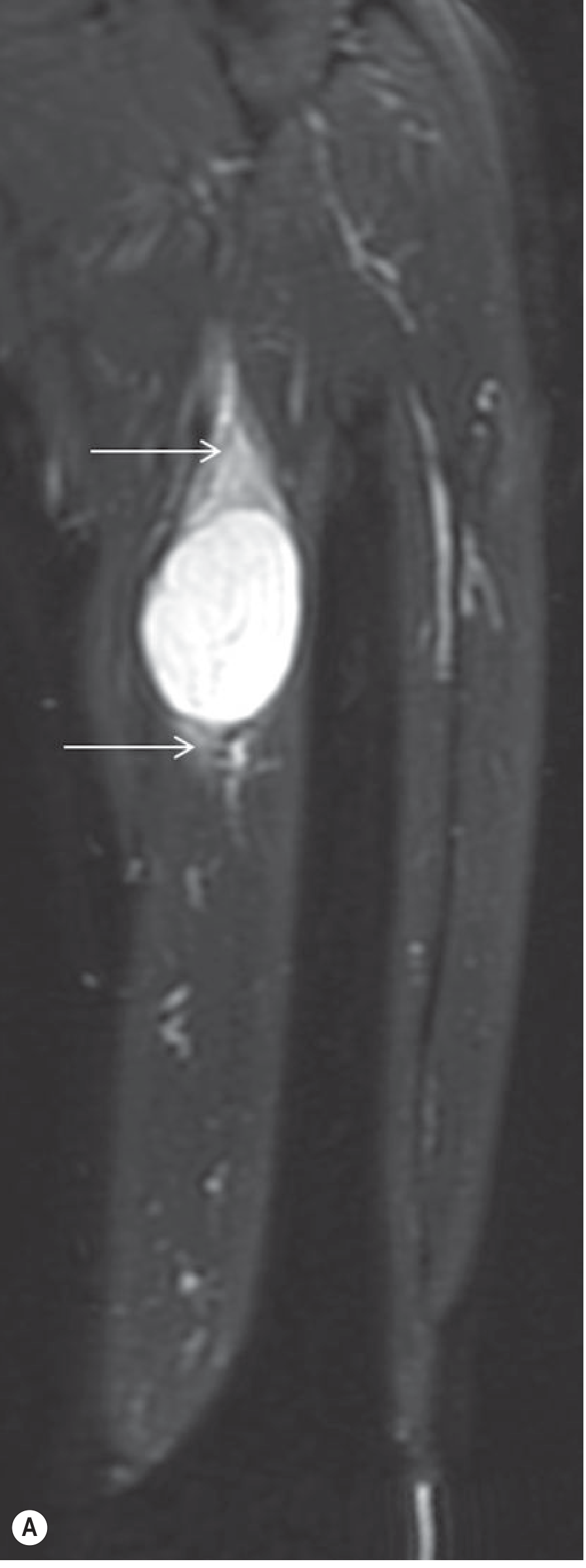
Intramuscular myxoma on MRI (the most useful modality):
- Low signal on T1
- Bright (fluid-like) signal on T2 - often resembles a cyst
- May contain thin fibrous septa and cystic areas
- Surrounding perilesional soft-tissue edema-like T2 signal ("bright rim" around the tumor)
- A rind of fat at the poles may simulate a "split-fat sign"
- Internal enhancement on contrast (confirms it is not purely cystic); more prominent enhancement in higher-cellularity tumors
- On ultrasound: hypoechoic ovoid mass with a hyperechoic rim ("bright rim sign")
Pathology
Fibrous dysplasia histology:
- Curvilinear trabeculae of woven bone with thin, irregular, C- and S-shaped spicules
- Absent osteoblastic rimming (a key diagnostic feature)
- Background of moderately cellular fibroblastic stroma with loose collagen
- Cystic degeneration, hemorrhage, and foamy macrophages may be present
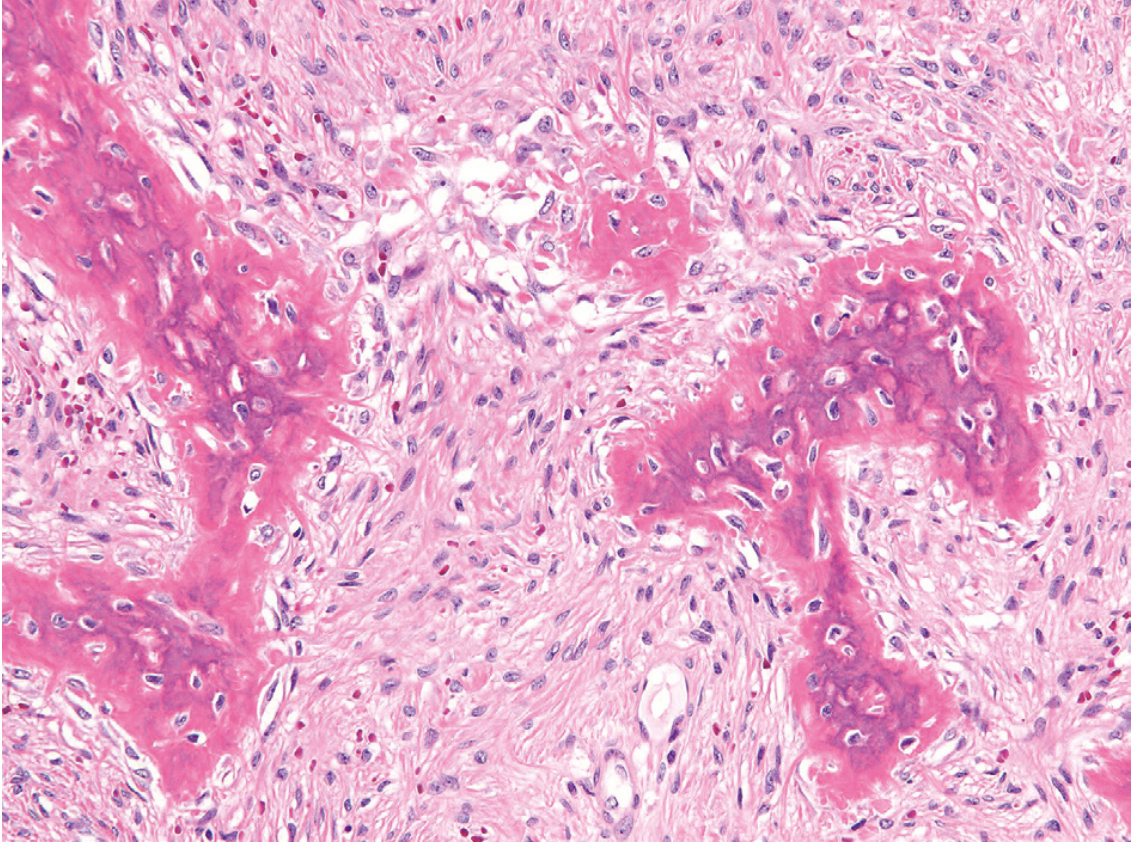
Intramuscular myxoma histology:
- Bland stellate and bipolar fibroblasts
- Abundant myxoid (mucoid) extracellular matrix
- Highly variable cellularity
- Histologically indistinguishable from sporadic intramuscular myxoma
The GNAS R201H mutation can be detected by molecular testing in myxoma tissue, which is diagnostically helpful in confirming Mazabraud syndrome.
Differential Diagnosis
| Feature | Mazabraud | McCune-Albright | Sporadic FD | Sporadic IM Myxoma |
|---|---|---|---|---|
| FD type | Usually polyostotic | Polyostotic | Mono or polyostotic | None |
| Myxomas | Yes (intramuscular) | No | No | Yes (sporadic) |
| Cafe-au-lait spots | Absent (unless MAS overlap) | Present | Absent | Absent |
| Endocrinopathy | Absent (unless MAS overlap) | Present | Absent | Absent |
| GNAS mutation | Yes (postzygotic) | Yes (postzygotic) | Yes (postzygotic) | Yes (postzygotic) |
The myxomas must be distinguished from:
- Myxofibrosarcoma / low-grade fibromyxoid sarcoma / myxoid liposarcoma - hypercellular areas of intramuscular myxoma can resemble these; however, sarcomas lack GNAS mutations
- Juxta-articular myxoma - predominantly pediatric, lacks GNAS mutation
- Cystic lesions (ganglia, synovial cysts, bursae) - distinguished by MRI enhancement
Treatment & Prognosis
No specific systemic therapy exists. Each component is managed on its own merits:
-
Fibrous dysplasia:
- Observation for asymptomatic lesions
- Curettage (and bone grafting if needed) for symptomatic lesions or impending pathologic fracture
- Surgical correction of deformity (e.g., intramedullary nailing for "shepherd's crook" femur)
- Bisphosphonates (e.g., pamidronate, zoledronic acid) for severe bone pain or extensive disease
- Malignant transformation to sarcoma is extremely rare but reported, especially in the polyostotic setting
-
Intramuscular myxomas:
- Observation if asymptomatic
- Surgical excision for compressive symptoms, pain, or limb deformity
- Recurrence after excision: ~30% at a median of 8.5 years (EMSOS study)
- Revision surgery required in ~25%
- High cellularity of myxomas is significantly associated with recurrence (p < 0.05)
- Despite local recurrence, malignant transformation of myxomas has not been documented
Overall prognosis is generally good. The syndrome is benign in the vast majority of cases, though the progressive nature of the bone disease and the tendency of myxomas to recur require long-term follow-up.
Key Sources
- Robbins, Cotran & Kumar - Pathologic Basis of Disease, pp. 1102-1103
- Grainger & Allison's Diagnostic Radiology, pp. 699, 2187-2191
- Campbell's Operative Orthopaedics 15th Ed, p. 1166
- Rheumatology, 2-Volume Set (Elsevier, 2022), p. 3990
- EMSOS multicenter study (PMID 30653046) - largest epidemiological dataset on Mazabraud syndrome (32 patients, 6 centers)
- PathologyOutlines.com - Mazabraud syndrome
This is a shared conversation. Sign in to Orris to start your own chat.