Describe multiple myeloma
Reading File
Finding Sources
Searching PubMed
"multiple myeloma"[MeSH Terms] AND treatment
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have all the information needed for a comprehensive answer. Here it is:
Multiple Myeloma
Multiple myeloma (MM) is a malignant neoplasm of clonal plasma cells that infiltrate the bone marrow and produce a monoclonal immunoglobulin (M protein). It is one of the most common lymphoid malignancies, accounting for ~1% of all cancers and slightly more than 10% of hematologic malignancies in the United States, with roughly 30,000 new cases per year.
Epidemiology
- Median age at diagnosis: ~65-70 years; only 2% of patients are younger than 40
- Sex: slightly more common in males
- Race: incidence in Black Americans is nearly twice that in White Americans (reason unknown)
- Almost all cases arise from a premalignant stage called monoclonal gammopathy of undetermined significance (MGUS), which progresses to myeloma at ~1% per year
Pathogenesis
The fundamental event is uncontrolled clonal expansion of terminally differentiated B cells (plasma cells). Key molecular mechanisms include:
- Chromosomal translocations involving the IgH locus on chromosome 14 - these commonly fuse IgH to oncogenes including cyclin D1 and cyclin D3, dysregulating D cyclins and driving cell proliferation
- IL-6 produced by bone marrow stromal fibroblasts and macrophages is the major growth and survival cytokine for myeloma cells
- MYC translocations appear late, especially in aggressive disease
- Myeloma cells upregulate RANKL on stromal cells, which activates osteoclasts, while simultaneously releasing factors that inhibit osteoblasts - the net result is unopposed bone resorption
The M protein produced is most commonly:
- IgG (~60%)
- IgA (~20-25%)
- Free light chains (kappa or lambda) only (~20%)
- IgM, IgD, IgE - rare
- ~1% are nonsecretory (no detectable M protein)
The "CRAB" Criteria (Myeloma-Defining Events)
The classic symptomatic features, which define active myeloma requiring treatment, are captured by the acronym CRAB:
| Letter | Feature | Mechanism |
|---|---|---|
| C | HyperCalcemia (>11 mg/dL) | Osteoclast-driven bone resorption |
| R | Renal insufficiency (creatinine >2 mg/dL) | Light chain cast nephropathy, hypercalcemia |
| A | Anemia (Hb <10 g/dL) | Marrow replacement by plasma cells |
| B | Bone lesions (lytic lesions, pathologic fractures) | RANKL-mediated osteoclast activation |
Additional myeloma-defining events include bone marrow plasma cells ≥60%, serum free light chain ratio ≥100, or >1 focal lesion on MRI.
Clinical Features
Skeletal
The most common presenting complaint is bone pain, typically in the back or chest. Lesions appear as sharply "punched-out" defects 1-4 cm in diameter, most commonly in the vertebral column, ribs, skull, pelvis, femur, clavicle, and scapula. Pathologic fractures are common, especially vertebral compression fractures.
Renal
Renal insufficiency is present in ~20% of patients at diagnosis and occurs in up to 50% overall. The main mechanisms are:
- Light chain cast nephropathy ("myeloma kidney"): Bence Jones proteins precipitate in distal tubules and collecting ducts, forming large waxy casts surrounded by multinucleate giant cells; tubular epithelium becomes necrotic
- Hypercalcemia: causes dehydration, renal vasoconstriction, and stone formation
- AL amyloidosis: (~10% of patients) - light chain deposits as amyloid in glomeruli and vessel walls, causing nephrotic syndrome
- Light chain deposition disease, Fanconi syndrome, bacterial pyelonephritis - additional contributors
Renal failure is the second leading cause of death (after infections).
Hematologic / Immune
- Normocytic normochromic anemia from marrow replacement; sometimes with leukopenia and thrombocytopenia
- Humoral immunodeficiency: myeloma cells suppress normal B-cell function; despite elevated total immunoglobulin (from M protein), production of functional antibodies is profoundly reduced - this predisposes patients to recurrent bacterial infections, which are the leading cause of death
Neurologic
- Radiculopathy (most common neurologic complication) - thoracic or lumbosacral nerve compression by vertebral lesion or collapsed bone
- Spinal cord compression in up to 10%
- Hypercalcemia-related: confusion, weakness, lethargy
- Peripheral neuropathy is uncommon and usually due to amyloidosis
Morphology
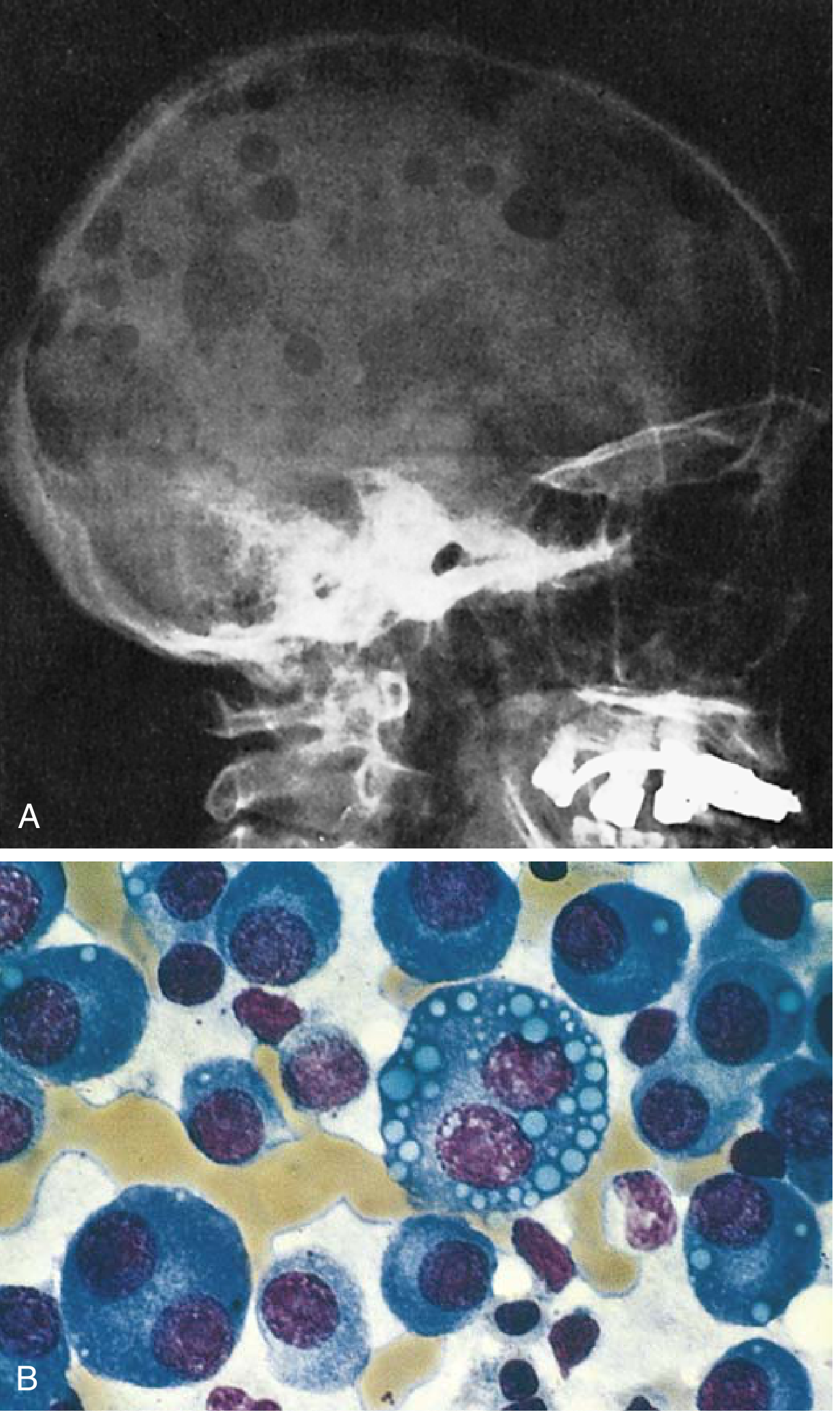
Fig. 10.28 Multiple myeloma. (A) Lateral skull radiograph showing classic sharply punched-out lytic lesions. (B) Bone marrow showing neoplastic plasma cells, some with cytoplasmic Russell bodies (immunoglobulin inclusions). - Robbins & Kumar Basic Pathology
- Bone marrow shows >10% clonal plasma cells (often >30% in active disease)
- Cells may resemble normal plasma cells or show abnormal features: prominent nucleoli, Russell bodies (cytoplasmic immunoglobulin inclusions)
- In advanced disease, plasma cells can spread to viscera and soft tissues; rarely, a plasma cell leukemia picture emerges (>5% plasma cells in peripheral blood or absolute count >500/μL)
Diagnosis
Diagnosis requires all three of:
- ≥10% clonal plasma cells on bone marrow biopsy
- M protein in serum or urine (or nonsecretory myeloma)
- Evidence of myeloma-defining events (CRAB criteria or biomarkers above)
Key investigations:
- Serum protein electrophoresis (SPEP): tall, narrow-based "M spike" in the gamma region
- Serum immunofixation: identifies the heavy and light chain class
- Serum free light chain assay: abnormal kappa/lambda ratio (normal 0.26-1.65); more sensitive than electrophoresis for free light chains
- 24-hour urine for Bence Jones proteins
- Bone marrow biopsy with flow cytometry and FISH (for cytogenetics)
- Imaging: whole-body low-dose CT, PET-CT, or MRI for bone lesions; classic plain films show punched-out lytic lesions ("raindrop skull")
- CBC, BMP (creatinine, calcium), LDH, beta-2 microglobulin (staging)
Differential from MGUS and smoldering myeloma:
- MGUS: <10% bone marrow plasma cells, M protein <3 g/dL, no end-organ damage
- Smoldering MM: 10-60% bone marrow plasma cells or M protein ≥3 g/dL, but no CRAB criteria or biomarker events - managed with observation unless high-risk
Staging
The Revised International Staging System (R-ISS) uses:
- Beta-2 microglobulin and albumin (ISS stages I-III)
- Cytogenetic abnormalities by FISH (high-risk: del 17p, t(4;14), t(14;16))
- LDH
Treatment
Newly Diagnosed, Transplant-Eligible
- Induction therapy: triplet or quadruplet regimens - typically bortezomib (proteasome inhibitor) + lenalidomide (immunomodulatory drug) + dexamethasone (VRd), now increasingly with daratumumab (anti-CD38 monoclonal antibody) added as a quadruplet (D-VRd)
- Autologous hematopoietic stem cell transplantation (ASCT): remains standard of care after induction; prolongs remission but is not curative
- Maintenance therapy: lenalidomide post-transplant extends progression-free survival
Newly Diagnosed, Transplant-Ineligible
- VRd or daratumumab-based combinations adapted for older/frailer patients
Relapsed/Refractory Disease
- Multiple salvage options: pomalidomide, carfilzomib, ixazomib, daratumumab, isatuximab, selinexor
- CAR-T cell therapy (e.g., idecabtagene vicleucel, ciltacabtagene autoleucel targeting BCMA) - approved for heavily pre-treated patients; recent meta-analyses (PMID 39551604) show CAR-T has superior outcomes vs. bispecific antibodies as third-line+ therapy
- Bispecific antibodies (e.g., teclistamab targeting BCMA×CD3) - newer agents with strong response rates
Supportive Care
- Bisphosphonates (zoledronic acid) or denosumab: reduce pathologic fractures and hypercalcemia
- Erythropoiesis-stimulating agents / transfusions for anemia
- Antimicrobial prophylaxis for infection risk
- Adequate hydration and management of hypercalcemia
Mechanism of Key Drug Classes
| Drug Class | Prototype | Mechanism |
|---|---|---|
| Proteasome inhibitors | Bortezomib, carfilzomib | Block proteasomal degradation of misfolded immunoglobulin chains, triggering apoptosis in plasma cells |
| IMiDs (immunomodulatory drugs) | Thalidomide, lenalidomide, pomalidomide | Bind cereblon (E3 ubiquitin ligase), stimulating degradation of pro-oncogenic proteins (Ikaros, Aiolos) in myeloma cells |
| Anti-CD38 mAbs | Daratumumab, isatuximab | Direct cytotoxicity against CD38-expressing plasma cells; also modulates immunosuppressive cells in the tumor microenvironment |
Prognosis
- Median survival: approximately 5 years (markedly improved from ~18 months in the pre-novel agent era)
- Patients with aggressive disease and multiple bony lesions, if untreated, rarely survive >6-12 months
- "Smoldering myeloma" may not require treatment for years
- Not currently curable with standard therapy; CAR-T and novel combinations are being studied for potential deeper, more durable remissions
- High-risk cytogenetics (del 17p, t(4;14), t(14;16)) confer significantly worse prognosis
Recent evidence note: A 2024 meta-analysis (PMID 39551604, J Immunother Cancer) found CAR-T cell therapy shows superior outcomes compared to bispecific antibodies in third-line or later treatment for multiple myeloma. CAR-T-associated secondary malignancy risk and non-relapse mortality are also being actively characterized in large systematic reviews (PMIDs 39256908, 38977912).
Sources: Robbins & Kumar Basic Pathology, p. 420-423 | Goldman-Cecil Medicine, International Edition, Chapter 173 | Robbins, Cotran & Kumar Pathologic Basis of Disease
Explain the pathophysiology of renal involvement (myeloma kidney) in detail
Finding Sources
Searching PubMed
"myeloma kidney" AND "light chain cast nephropathy" AND pathophysiology
Finding Sources
Searching PubMed
"multiple myeloma" AND "renal" AND "light chain"
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
I now have everything needed for a thorough, mechanistically detailed answer. Here it is:
Pathophysiology of Renal Involvement in Multiple Myeloma ("Myeloma Kidney")
Renal insufficiency is one of the most serious complications of multiple myeloma, occurring in approximately 20% of patients at diagnosis and up to 50% overall. It is the second leading cause of death after infections. The term "myeloma kidney" specifically refers to light chain cast nephropathy (LCCN), though renal injury in myeloma is multifactorial and involves several distinct mechanisms operating simultaneously or sequentially.
Overview of Mechanisms
| Mechanism | Predominant Site | Light Chain Type | Clinical Presentation |
|---|---|---|---|
| Light chain cast nephropathy | Distal tubule / collecting duct | Any (cast-forming) | AKI or progressive CKD |
| Proximal tubule direct toxicity | Proximal tubule | Any | Fanconi syndrome, isolated RTA |
| AL amyloidosis | Glomeruli / vessels | Usually λ (lambda) | Nephrotic syndrome |
| Light chain deposition disease (LCDD) | GBM / mesangium / tubular BM | Usually κ (kappa) | Nephrotic syndrome + CKD |
| Hypercalcemia | Tubules / glomeruli | - | AKI, nephrocalcinosis |
| Hyperuricemia | Collecting ducts | - | Obstructive AKI (tumor lysis) |
1. Light Chain Cast Nephropathy - The Primary Mechanism
This is the dominant cause of acute renal failure in myeloma. The sequence of events is as follows:
Step 1: Overflow of Free Light Chains into the Filtrate
Normally, the kidneys freely filter low-molecular-weight proteins, including immunoglobulin free light chains (FLCs, ~22 kDa). In myeloma, the massive overproduction of monoclonal FLCs by the plasma cell clone overwhelms the normal route of tubular catabolism. Once the serum FLC concentration is very high (typically >150 mg/dL in cast nephropathy), the proximal tubule's reabsorptive capacity is saturated and large quantities of light chains spill into the distal nephron as Bence Jones proteins in the urine.
Step 2: Proximal Tubule Saturation and Toxicity
Under normal conditions, filtered FLCs are reabsorbed in the proximal tubule by binding to the megalin-cubilin heterodimeric receptor complex on the brush border. Once internalized:
- They are trafficked to endosomes and lysosomes for catabolism
- Accumulation in lysosomes causes cellular vacuolization, desquamation, and loss of brush border
- Certain light chains generate hydrogen peroxide, which triggers NF-κB activation and production of inflammatory cytokines (including MCP-1, a monocyte chemoattractant)
- FLCs promote apoptosis via apoptosis signal-regulating kinase 1 (ASK1/MAP3K5)
- These events drive tubulointerstitial inflammation and fibrosis
Sublethal proximal tubule injury produces Fanconi syndrome - a full proximal tubular defect with glucosuria, phosphaturia, aminoaciduria, and sometimes renal tubular acidosis (RTA), even without overt cast nephropathy.
Step 3: Cast Formation in the Distal Nephron
Once the proximal tubule's reabsorptive capacity is exceeded, a high concentration of FLCs reaches the distal tubule and collecting duct - the site where cast nephropathy begins. The key co-participant is Tamm-Horsfall protein (THP), also called uromodulin:
- THP is synthesized exclusively by cells of the thick ascending limb of the loop of Henle and is the most abundant protein in normal urine
- Cast-forming light chains bind to a specific site on the peptide backbone of THP via the CDR3 (complementarity-determining region 3) domain of the light chain
- This binding causes co-aggregation (co-precipitation) of the FLC-THP complex, occluding the tubular lumen
- The resulting casts are large, waxy, laminated, often concentrically layered and fractured, and stain pink to blue on H&E
Critical insight: Not all light chains are equally cast-forming. The binding affinity is determined by the CDR3 domain's physicochemical properties - some light chains co-precipitate readily with THP, while others do not, which explains why not every patient with Bence Jones proteinuria develops cast nephropathy.
Step 4: Factors That Accelerate Cast Formation
The co-precipitation is highly sensitive to the ionic environment and tubular flow rate:
- Volume depletion - reduces tubular flow, extending contact time between FLCs and THP in the distal tubule; can convert previously non-cast-forming light chains into toxic ones
- Hypercalcemia and high NaCl concentration - directly facilitate THP-FLC co-aggregation
- Acidic urine - promotes precipitation (alkalinization reduces it)
- Low GFR (from any cause: NSAIDs, contrast agents, infection, dehydration) - reduces filtration but paradoxically concentrates remaining light chains distally
- Furosemide - increases distal Cl⁻ concentration, which augments THP secretion and accelerates FLC co-precipitation; loop diuretics can actually worsen cast nephropathy and should be used cautiously
Step 5: Tubular Obstruction and Inflammatory Consequences
Once casts occlude the tubular lumen:
- Intranephron pressure rises proximal to the obstruction, simultaneously reducing single-nephron GFR
- The obstruction provokes a foreign-body giant cell reaction: activated macrophages surround the casts as multinucleated giant cells
- Adjacent tubular epithelial cells undergo necrosis and atrophy
- The interstitium develops mononuclear cell infiltration and progressive fibrosis
- Casts can rupture the tubular wall, evoking a granulomatous reaction in the peritubular interstitium
- Interstitial fibrosis can develop rapidly, making early treatment critical to preserve renal function
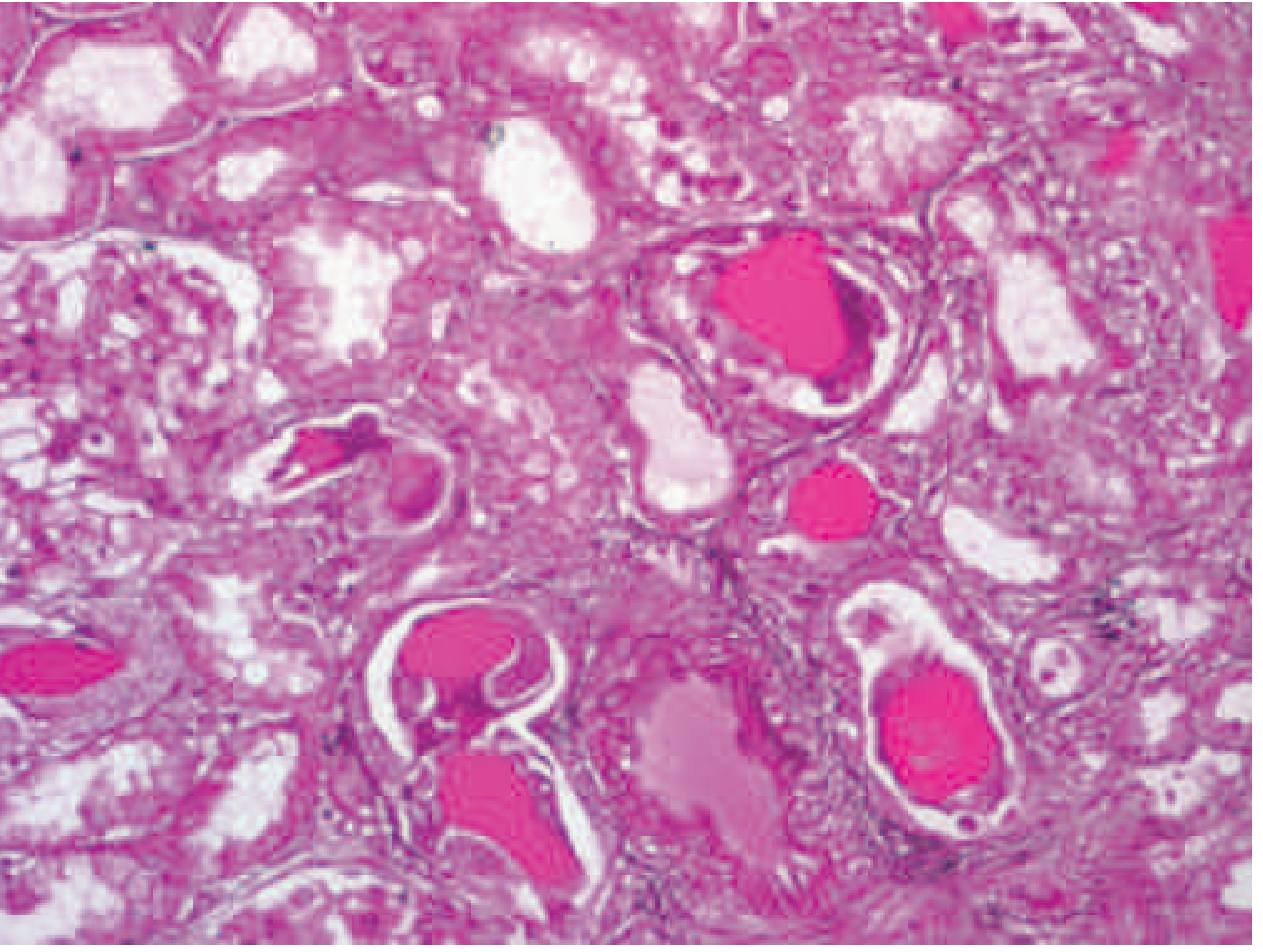
Histologic appearance of myeloma cast nephropathy. H&E-stained kidney biopsy showing atrophic tubules filled with eosinophilic Bence Jones protein casts, surrounded by characteristic giant cell reactions. Glomeruli are typically normal. - Harrison's Principles of Internal Medicine, 22nd Ed.
2. Hypercalcemia-Mediated Renal Injury
Hypercalcemia is present at diagnosis in 15-20% of myeloma patients and acts through multiple converging mechanisms:
- Renal vasoconstriction - reduces GFR directly
- Decreased glomerular ultrafiltration coefficient - impairs filtration
- Nephrogenic diabetes insipidus - calcium deposits around basement membranes of distal tubules and collecting ducts, producing a vasopressin-resistant concentrating defect; leads to polyuria and paradoxical volume depletion, which in turn accelerates cast formation
- Nephrocalcinosis - calcium deposition triggers mononuclear cell infiltration and tubular death
- Renal stone formation (calcium-based) - can cause obstructive uropathy
Hypercalcemia and volume depletion are the two most common reversible precipitants of acute-on-chronic renal failure in myeloma.
3. AL Amyloidosis
- Occurs in approximately 6-24% of myeloma patients (some sources cite ~10%)
- Almost exclusively involves lambda (λ) light chains
- Misfolded lambda FLCs aggregate into β-pleated sheet fibrils (~8-10 nm diameter) that deposit in the glomerular mesangium, glomerular basement membrane, and vessel walls
- Deposits are Congo-red positive (apple-green birefringence under polarized light) and thioflavin T positive
- Clinical presentation: nephrotic-range proteinuria (predominantly albuminuria, not just Bence Jones), with or without renal insufficiency
- Amyloid deposits also compromise tubular function, exacerbate interstitial disease, and can deposit in myocardium, liver, peripheral nerves, and soft tissues
4. Light Chain Deposition Disease (LCDD)
- Less common than AL amyloidosis
- Almost exclusively involves kappa (κ) light chains (in contrast to amyloidosis)
- Light chains deposit as non-fibrillar, granular, electron-dense deposits along the glomerular basement membrane (GBM), mesangium, and tubular basement membranes
- Unlike amyloid, these deposits are Congo-red negative
- Tubular basement membrane involvement causes tubulointerstitial nephritis
- Clinically: nephrotic syndrome plus progressive CKD; can present as nodular glomerulopathy resembling diabetic nephropathy
5. Hyperuricemia
- Results from high tumor cell turnover, especially after chemotherapy (tumor lysis)
- Uric acid crystallizes in the collecting ducts and renal pelvis, causing obstructive AKI (acute urate nephropathy)
- Prevented prophylactically with allopurinol or rasburicase before initiating chemotherapy
6. Acquired Fanconi Syndrome
A distinct proximal tubular syndrome (independent of cast nephropathy) characterized by:
- Glucosuria (with normal plasma glucose)
- Phosphaturia - leading to hypophosphatemia and osteomalacia
- Aminoaciduria
- Renal tubular acidosis (RTA), type 2 (proximal)
- Caused by specific light chains (usually κ) that form intracellular crystals in proximal tubule lysosomes, impairing transport of glucose, amino acids, and phosphate
Synergistic Worsening
In practice, these mechanisms do not act in isolation. A typical patient trajectory:
- Myeloma produces large quantities of monoclonal FLCs → Bence Jones proteinuria
- Concurrent hypercalcemia causes volume depletion and renal vasoconstriction → GFR falls
- Reduced GFR concentrates light chains in the distal tubule → cast formation accelerated
- An intercurrent infection, NSAID use, or IV contrast further reduces GFR → acute-on-chronic injury
- Giant cell reaction → interstitial fibrosis → irreversible nephron loss if untreated
Clinical Clues to Diagnosis
- Proteinuria detected by sulfosalicylic acid (SSA) or urine protein electrophoresis but negative dipstick (dipstick detects albumin only, not light chains)
- Narrowed anion gap - due to cationic M protein (IgG or IgA) shifting the anion gap calculation
- Serum FLC ratio markedly abnormal; FLC level >500 mg/L strongly suggests cast nephropathy
- Bence Jones proteinuria on 24-hour urine immunofixation
- Kidney biopsy: diagnostic - characteristic casts with giant cell reaction; glomeruli are usually normal in appearance in pure LCCN (distinguishing from glomerular diseases like amyloidosis or LCDD)
Principles of Treatment
- Treat the plasma cell clone aggressively to rapidly reduce circulating FLC levels - bortezomib-based regimens are preferred (bortezomib/cyclophosphamide/dexamethasone ± daratumumab); rapid FLC reduction is the single most important determinant of renal recovery
- Aggressive hydration to maintain urine output ~100 mL/hour, which dilutes light chains in the tubule and reduces contact time with THP
- Avoid furosemide acutely - it worsens FLC-THP co-aggregation by increasing distal Cl⁻
- Correct hypercalcemia with IV fluids, bisphosphonates, calcitonin
- Avoid nephrotoxins: NSAIDs, IV contrast (use iso-osmolar agents if essential), aminoglycosides
- Alkalinize urine (sodium bicarbonate) - reduces FLC-THP precipitation
- Allopurinol for hyperuricemia
- Hemodialysis for symptomatic azotemia
- High-cutoff hemodialysis (HCHD) to remove circulating FLCs - mixed trial results (EuLITE showed no benefit; MYRE showed benefit at 6 and 12 months but not primary endpoint); not standard of care
- Plasmapheresis - recommended by Goldman-Cecil when FLC >150 mg/dL, daily for 5 days; however, broader evidence remains equivocal
Recent evidence note (PMID 39662762, Presse Med 2025): A comprehensive 2025 review by Bridoux, Leung, and Nasr highlighted the importance of early renal biopsy in myeloma-associated AKI to distinguish LCCN from other myeloma-related nephropathies (amyloidosis, LCDD, MGRS-associated lesions), as management differs significantly between them. The 2025 Renal Pathology Society/IKMG consensus (PMID 40280412) has also standardized pathologic definitions for monoclonal gammopathy-associated kidney lesions.
Sources: Brenner & Rector's The Kidney, 2-Volume Set | NKF Primer on Kidney Diseases, 8th Ed. | Robbins, Cotran & Kumar Pathologic Basis of Disease | Goldman-Cecil Medicine, Chapter 173 | Harrison's Principles of Internal Medicine, 22nd Ed., Chapter 328
This is a shared conversation. Sign in to Orris to start your own chat.