Stress fracture etiology pathology signs and symptoms investigation and treatment of this explain in detail with appropriate illustrative images and diagrams
Reading File
Finding Sources
Finding Sources
Searching Images
stress fracture bone X-ray MRI
Reading File
Reading File
Reading File
Searching Images
stress fracture pathophysiology bone remodeling mechanism
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
I now have all the material needed to compile a comprehensive, well-illustrated answer. Let me compose the full response.
Stress Fractures: A Comprehensive Review
1. Definition & Historical Perspective
A stress fracture is a fatigue failure of bone resulting from the accumulation of microdamage caused by repeated sub-maximal loading — loads that individually would not fracture the bone but collectively exceed the bone's reparative capacity. This distinguishes it from:
- Fatigue fracture: abnormal repetitive stress applied to normal bone (e.g., military recruits, athletes)
- Insufficiency fracture: normal/physiological stress applied to abnormal (weakened) bone (e.g., osteoporosis)
First described by Breithaupt in Prussian military recruits in 1855, they were historically called "march fractures" because of their prevalence among soldiers undergoing prolonged marching.
— Rockwood and Green's Fractures in Adults 10th ed., p. 851
2. Etiology & Risk Factors
Intrinsic (Patient) Factors
| Category | Specific Factors |
|---|---|
| Sex | Twice as common in women vs. men |
| Hormonal status | Amenorrhea, oligomenorrhea, female athlete triad |
| Nutritional | Calcium/vitamin D deficiency, eating disorders, negative energy balance |
| Bone geometry | Narrower bone cross-section, lower bone density |
| Race | Lower baseline BMD in White women (highest risk group) |
| Age | Elderly (osteoporosis-related) and adolescents (growth-related) |
| Neuromuscular | Poor shock absorption, muscle fatigue, biomechanical malalignment |
| Genetics | Polymorphisms affecting bone density and remodeling |
Extrinsic (Environmental) Factors
- Abrupt increase in training volume, intensity, or frequency ("too much, too fast")
- Hard training surfaces
- Worn-out footwear; inadequate equipment
- Poor technique / biomechanics
- Rapid transition in activity type (e.g., running on asphalt after turf)
The Female Athlete Triad
Female athletes with eating disorders + amenorrhea/oligomenorrhea + low bone density are at exceptionally high risk — the triad disrupts the hormonal and nutritional milieu required for bone remodeling.
— Textbook of Family Medicine 9e, p. 800; Rockwood and Green's, p. 852
3. Pathophysiology
Bone as a Material: Fatigue Failure in Three Stages
Stress fractures represent material fatigue failure of bone — the same physical principle that causes metal to crack under repeated bending:
Stage 1 — Crack Initiation
- Every loading episode places strain on bone, causing some microdamage
- Microcrack initiation occurs at sites of stress concentration (lacunae, canaliculi, cement lines)
- Alone, this is not pathological — it is the first step in normal bone remodeling
- Bone metabolic units (BMUs / "cutting cones") repair these microcracks constantly
Stage 2 — Crack Propagation
- If loading frequency or intensity exceeds the repair rate, cracks propagate
- Propagation occurs preferentially along cement lines (faster when parallel to them than perpendicular)
- Multiple microcracks coalesce → symptomatic stress fracture
Stage 3 — Complete Fracture
- Without activity modification, structural failure occurs
"Since in vitro bone appears to have no endurance limit, with continued loading, microdamage will continue to accumulate until complete fracture occurs." — Rockwood and Green's, p. 851
Wolff's Law & Healthy Remodeling
Under normal circumstances, loaded bone adapts by becoming stronger (Wolff's Law). The balance between microcrack creation and repair maintains bone health. When this balance is disrupted (overload or impaired healing), a stress fracture results.
The Neuromuscular Hypothesis
Muscles have a dual role:
- Provocative: muscle contraction generates compressive, tensile, and rotational internal stresses on bone
- Protective: well-conditioned muscles distribute external loads, acting as shock absorbers
Muscle fatigue during prolonged exercise shifts more load onto bone, raising stress fracture risk.
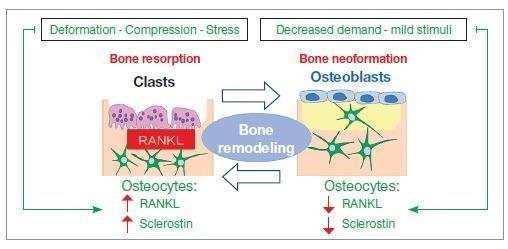
Bone Remodeling Pathway
The diagram below illustrates how mechanical stress drives osteocyte-mediated signaling through RANKL and sclerostin to modulate osteoblast/osteoclast balance:
4. Common Locations
The tibia accounts for ~50% of all stress fractures in running and jumping athletes. Distribution by site:
| Site | Proportion / Population |
|---|---|
| Tibia (mid-distal shaft) | ~50%; runners |
| Tibia (proximal third) | Adolescents |
| Metatarsals (2nd > 3rd) | Military recruits, dancers ("march fracture") |
| Fibula (distal) | Runners |
| Femoral neck | Long-distance runners (HIGH RISK) |
| Navicular | Sprinters, high jumpers (HIGH RISK) |
| Femoral shaft | Distance runners |
| Sacrum/Pelvis | Elderly, osteoporotic patients |
| Ribs | Rowers, throwing athletes |
| Humerus/Olecranon | Baseball pitchers, gymnasts |
5. Signs & Symptoms
Symptom Progression (Characteristic Pattern)
- Early: Pain only during activity → relieves completely with rest
- Intermediate: Pain persists for hours after activity, forcing the athlete to stop
- Late: Pain with walking and eventually at rest
"Pain from a stress fracture begins with mild pain during activity that resolves with rest. As the stress fracture progresses, pain increases during activity and continues for hours afterward." — Textbook of Family Medicine 9e, p. 800
Clinical Examination Findings
| Sign | Description |
|---|---|
| Point tenderness | Focal, reproducible tenderness on direct palpation over the fracture site |
| Local swelling | Variable; may or may not be present |
| Periosteal thickening | Palpable in later stages |
| Fulcrum test | Long bone (femur/tibia): examiner's forearm acts as fulcrum; pain reproduction indicates stress fracture |
| Single-leg hop test | Painful hopping on the affected limb; patient may refuse to hop — useful for lower extremity stress fractures |
| Tuning fork test | Vibration placed over the fracture site increases pain (sensitivity ~50–70%) |
| Load/percussion test | Percussion along bone axis causes focal pain |
"Physical examination reveals reproducible point tenderness with direct palpation of the affected bone site." — Rockwood and Green's, p. 855
6. Classification
Kaeding–Miller Stress Fracture Classification System
(The most widely used clinical grading system)
| Grade | Pain | Imaging Findings |
|---|---|---|
| I | None | Imaging evidence of stress fracture; no fracture line |
| II | Present | Imaging evidence; no fracture line |
| III | Present | Non-displaced fracture line |
| IV | Present | Displaced fracture (>2 mm) |
| V | Present | Nonunion |
Low-Risk vs. High-Risk Stress Fractures
Low-risk sites (compressive side, favorable healing):
- Femoral shaft, medial tibia, ribs, ulnar shaft, metatarsals 1–4
- Respond well to activity modification
- Unlikely to progress to nonunion
High-risk sites (tension side, prone to nonunion/complete fracture):
- Femoral neck (tension side) — risk of avascular necrosis
- Anterior tibial cortex — "dreaded black line" on lateral X-ray
- Tarsal navicular — poor blood supply
- 5th metatarsal diaphysis (Jones fracture zone)
- Medial malleolus
- Sesamoids of the hallux
- Olecranon (in throwers)
"High-risk stress fractures tend to progress to nonunion or complete fracture, require operative management, and recur." — Rockwood and Green's, p. 858
7. Investigations
Plain Radiographs (X-ray)
- Sensitivity: ~50% overall; only 10–15% in the first 2 weeks
- Two-thirds of initial X-rays are negative
- Findings when positive:
- Periosteal reaction / new bone formation (most common)
- Sclerotic line (cortical thickening at the fracture site)
- "Dreaded black line" — radiolucent transverse line through anterior tibial cortex (high-risk sign)
- Endosteal thickening
- Repeat X-ray 10–14 days later may show periosteal elevation or demineralization as healing begins
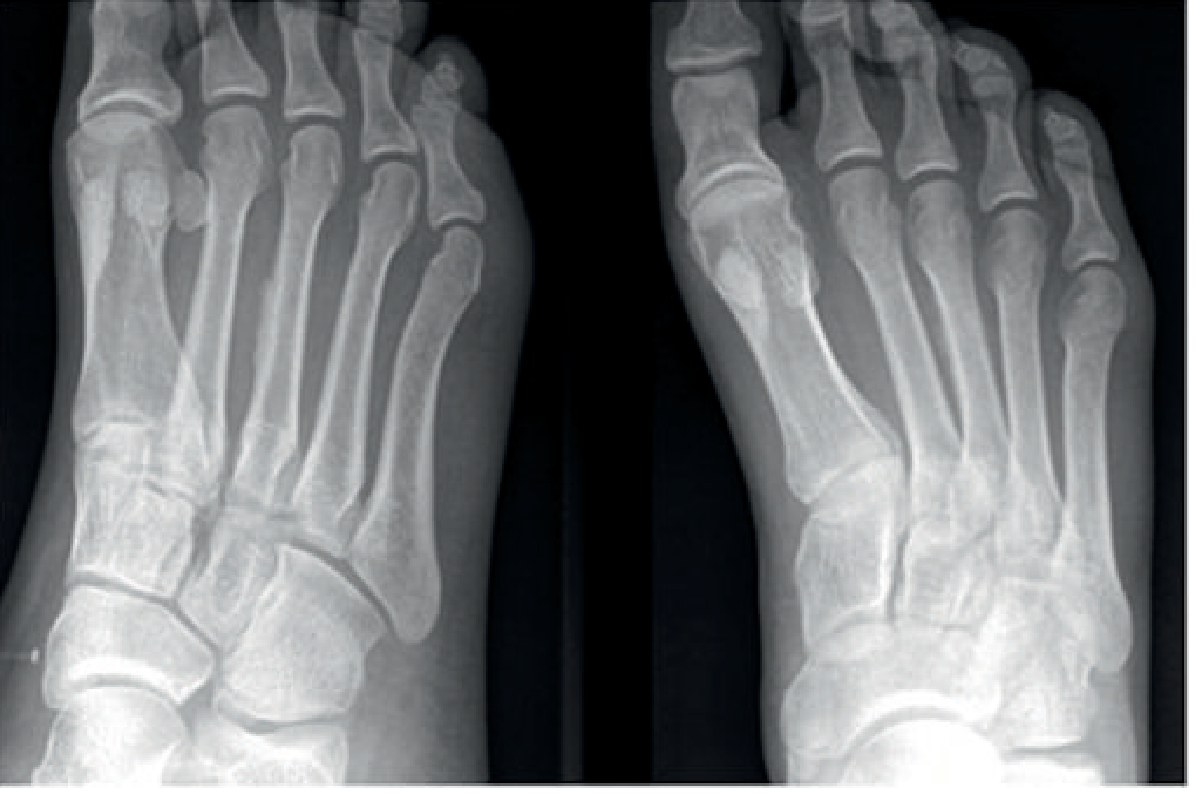
Fig: Periosteal reaction around the 3rd metatarsal shaft bilaterally — a classic radiographic sign of a healing stress fracture. (Grainger & Allison's Diagnostic Radiology)
Bone Scintigraphy (Technetium-99m Bone Scan)
- Sensitivity: ~100% — positive 1–2 weeks before X-ray changes
- Lower specificity than MRI
- Triple-phase scan (angiogram + blood pool + delayed): stress fractures positive in all three phases; periostitis (shin splints) positive only in delayed phase and has a diffuse linear pattern vs. the focal "hot spot" of a stress fracture
- Limitation: uptake remains elevated for 12–18 months, lagging behind clinical resolution → less useful for return-to-sport decisions
MRI (Gold Standard)
- Sensitivity: ~99%; Specificity: ~>85%
- Modality of choice — detects bone marrow edema before X-ray changes
- STIR/T2 sequences: high signal (bright) in bone marrow edema at fracture site
- T1: low signal (dark) fracture line within edema
- Fredericson MRI Grading (tibia):
- Grade 1: periosteal edema on STIR
- Grade 2: +marrow edema on T2 but not T1
- Grade 3: +marrow edema on both T1 and T2
- Grade 4: fracture line visible
- MRI also assesses extent, guides return-to-sport timing, and identifies soft tissue injury
Multi-modality imaging comparison:
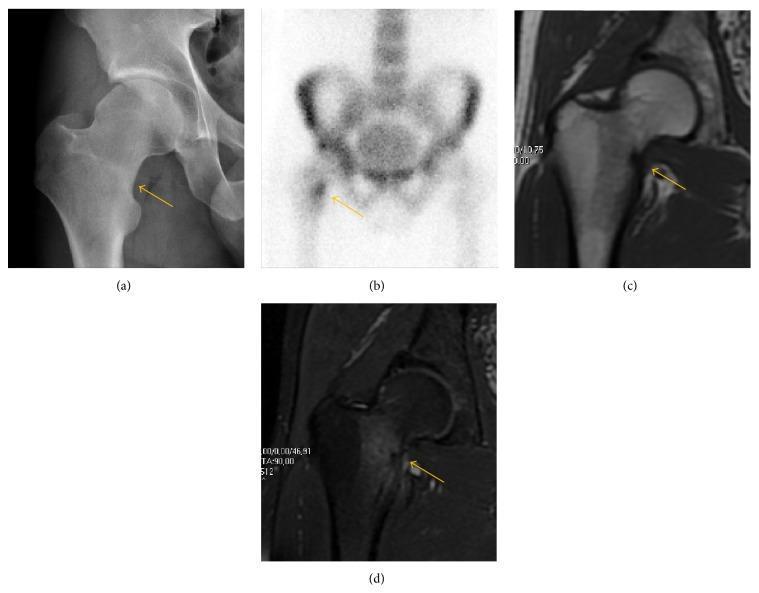
Fig: Femoral neck stress fracture — (a) subtle sclerotic line on AP X-ray, (b) focal radionuclide uptake on bone scan, (c) T1 MRI with hypointense fracture line, (d) fat-saturated proton density MRI demonstrating bright marrow edema
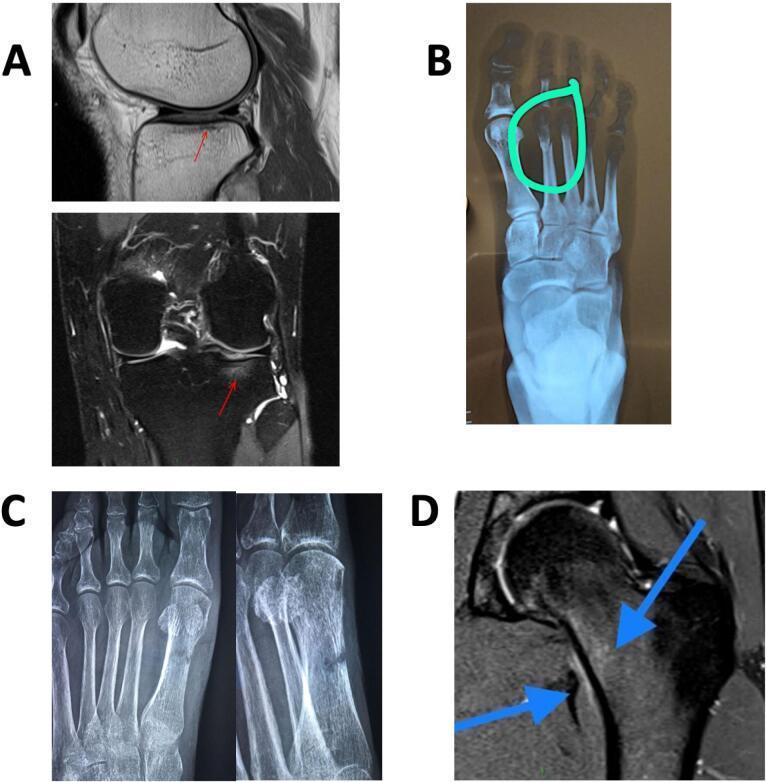
Fig: Multi-site stress fractures across modalities — tibial plateau on MRI (A), 2nd metatarsal X-ray (B), metatarsal healing with cortical thickening (C), femoral neck MRI marrow edema (D)
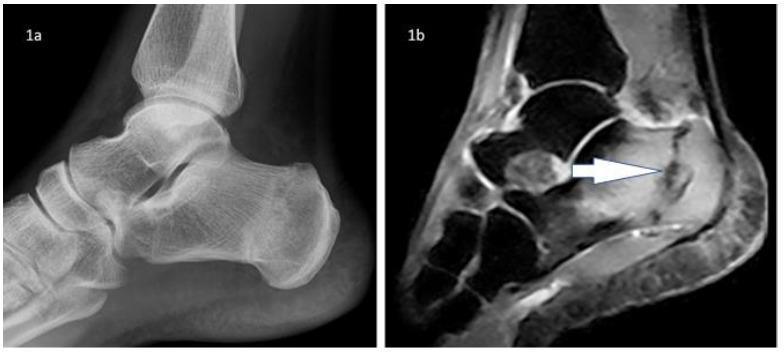
Fig: Calcaneal stress fracture — lateral X-ray (subtle sclerosis) vs. STIR MRI (clear linear fracture line with surrounding marrow edema)
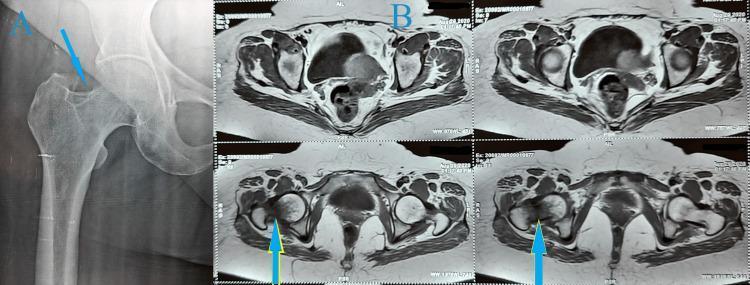
Fig: Femoral neck stress fracture — AP X-ray shows subtle sclerosis; axial MRI confirms marrow edema and fracture line (HIGH RISK — requires urgent orthopedic referral)
CT Scan
- Useful for precise characterization of complex fractures (tarsal navicular, sacrum)
- Identifies cortical disruption and displaced fragments
- Less sensitive than MRI for early/bone-marrow-only lesions
Ultrasound
- Limited reliability; not routinely recommended for diagnosis of bone stress injuries
SPECT (Single-Photon Emission CT)
- Combines bone scan sensitivity with anatomical localization; useful for complex pelvic/sacral fractures
Laboratory Investigations (Selected Patients)
- DEXA scan: patients with ≥2 stress fractures, female athletes with menstrual irregularity
- Vitamin D (25-OH-D), calcium, phosphate, PTH
- CBC, ESR/CRP: to exclude infection or malignancy
- Thyroid function tests (if metabolic bone disease suspected)
- Serum protein electrophoresis (exclude myeloma in older patients)
8. Treatment
General Principles
Treatment is individualized based on:
- Site (high-risk vs. low-risk)
- Grade (Kaeding–Miller I–V)
- Activity level / occupation of the patient
- Underlying risk factors (nutrition, hormonal status, bone density)
Low-Risk Stress Fractures — Conservative Management
| Step | Details |
|---|---|
| Activity modification | Immediate discontinuation of causative activity |
| Pain-free ambulation | Patient must be able to walk without pain |
| Crutches | Non-weight-bearing if walking is painful |
| Immobilization | Pneumatic leg brace for tibial fractures; rigid walking boot for foot fractures |
| Cross-training | Swimming, cycling, pool running — maintain fitness without impact |
| Analgesia | NSAIDs (short-term); consider avoiding NSAIDs long-term as they may impair bone healing |
| Nutrition | Calcium 1000–1500 mg/day; Vitamin D 800–1000 IU/day |
| Footwear | Shock-absorbing insoles, appropriate footwear |
| Healing time | 4–12 weeks depending on site and grade |
"Ambulation must be pain free to allow for fracture healing. If the athlete cannot achieve pain-free ambulation, a period of non-weight bearing on crutches is indicated." — Textbook of Family Medicine 9e, p. 801
High-Risk Stress Fractures — Aggressive Management
| Site | Management |
|---|---|
| Femoral neck (tension side) | Non-weight bearing + urgent surgical fixation (screw fixation) |
| Femoral neck (compression side, non-displaced) | Non-weight bearing; surgical fixation if at risk for displacement |
| Anterior tibial cortex | Non-weight bearing cast; surgical intramedullary nail fixation if "dreaded black line" or non-healing |
| Tarsal navicular | Non-weight bearing cast × 6–8 weeks; surgical fixation if displaced or non-union |
| 5th metatarsal (Jones fracture zone) | Non-weight bearing cast; early screw fixation in athletes |
| Medial malleolus | Surgical fixation (screw) to prevent progression |
| Olecranon (throwing athletes) | Surgical fixation |
"Treatment decision making for high-risk stress fractures should be based on radiographic findings with less consideration given to symptom severity." — Rockwood and Green's, p. 858
Grading-Based Treatment Summary
| Kaeding–Miller Grade | Treatment |
|---|---|
| I (no pain) | Observation; activity modification as tolerated |
| II (pain, no fracture line) | Activity restriction; non-impact cross-training |
| III (non-displaced fracture line) | Protected weight bearing / non-weight bearing + brace or cast |
| IV (displaced) | Surgical fixation in most cases |
| V (nonunion) | Surgical fixation ± bone grafting |
Return to Activity
- Gradual, pain-guided progression after clinical and radiographic healing
- Return to sport after pain-free activity at each level for at least 1 week
- Bone scan normalization takes 12–18 months but is not required before return to sport
- MRI resolution of edema is more reliable for guiding return
Addressing Underlying Risk Factors
- Female athlete triad: multidisciplinary management — dietitian, physician, sports psychologist; restore menstrual function
- Osteopenia/osteoporosis: DEXA, vitamin D/calcium supplementation, consider bisphosphonates in postmenopausal patients
- Biomechanical correction: orthotics for flat/high-arched feet; gait retraining
- Training modification: gradual progressive loading ("10% rule")
Emerging & Adjunct Therapies
- Low-intensity pulsed ultrasound (LIPUS): may accelerate healing in non-union cases
- Extracorporeal shock wave therapy (ESWT): for recalcitrant cases
- Bone stimulators (electromagnetic): evidence limited but used in non-union
- Bisphosphonates: caution — may paradoxically cause atypical femoral stress fractures with long-term use
9. Prevention
| Strategy | Mechanism |
|---|---|
| Gradual training progression | Allows bone remodeling to keep pace with loading |
| Adequate calcium and vitamin D | Optimizes bone mineral density |
| Appropriate footwear | Reduces shock transmission |
| Biomechanical screening | Identifies gait abnormalities |
| Cross-training | Distributes load across muscle groups |
| Pre-participation screening | Identifies at-risk athletes (triad, low BMD) |
| Shock-absorbing surfaces | Reduces peak forces |
10. Summary Flow
Repetitive Sub-maximal Loading
↓
Microcrack Initiation (normal)
↓
Overload > Repair Rate
↓
Crack Propagation → Coalescence
↓
Symptomatic Stress Fracture
↓
┌─────────────────────┐
│ │
Low-Risk Site High-Risk Site
│ │
Conservative Rx NWB ± Surgery
(rest, brace, (urgent fixation
cross-training) for displacement)
│ │
└─────────┬───────────┘
↓
Graduated Return to Activity
Key Sources:
- Rockwood and Green's Fractures in Adults, 10th ed. (2025), Ch. 24 — Stress Fractures
- Textbook of Family Medicine, 9th ed., Ch. 29 — Sports Medicine
- Tintinalli's Emergency Medicine, Ch. 277 — Leg Injuries
- Grainger & Allison's Diagnostic Radiology, Ch. 45 — Lower Limb
- Bailey and Love's Short Practice of Surgery, 28th ed., Ch. 5 — Foot
Sickle cell disease basics and it's relation to Orthopedic problems their associated signs and symptoms and investigations and evaluation treatment explain in detail with appropriate illustrative diagrams and images
Finding Sources
Finding Sources
Searching Images
sickle cell disease pathophysiology red blood cell sickling
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Searching Images
sickle cell osteonecrosis avascular necrosis femoral head X-ray MRI
Reading File
Reading File
Loading Image
Loading Image
Reading File
Finding Sources
Loading Image
Loading Image
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
Loading Image
I now have all the material needed for a comprehensive, well-illustrated response. Let me compose it.
Sickle Cell Disease: Basics & Orthopaedic Complications
Part I — Basics of Sickle Cell Disease
1. Definition & Genetics
Sickle Cell Disease (SCD) is a group of autosomal recessive haemoglobinopathies caused by a point mutation in the β-globin gene on chromosome 11. The substitution of valine for glutamic acid at position 6 of the β-globin chain produces haemoglobin S (HbS). Under deoxygenated conditions, HbS polymerises into long rigid rods, distorting the red cell into a characteristic sickle shape.
Genotypes and clinical severity:
| Genotype | Condition | Severity |
|---|---|---|
| HbSS | Sickle cell anaemia | Most severe |
| HbSC | Sickle-haemoglobin C disease | Moderate; paradoxically more bone infarction |
| HbS-β-thal | Sickle-β-thalassaemia | Can equal HbSS severity |
| HbSA | Sickle cell trait | Usually asymptomatic; rare crises |
"If the second β-chain is also HbS, then the patient has homozygous HbS-S, defined as sickle cell anaemia." — Grainger & Allison's Diagnostic Radiology, p. 1717
2. Epidemiology
- Primarily affects people of sub-Saharan African origin; also Middle East, Mediterranean, India
- ~8–10% of Black Americans carry sickle cell trait; ~0.2% have sickle cell anaemia
- Homozygous SCD reduces average life expectancy by 25–30 years (most patients die by age 50)
- The HbS gene confers relative protection against Plasmodium falciparum malaria — explaining its high frequency in malaria-endemic regions
3. Pathophysiology
Step-by-step mechanism:
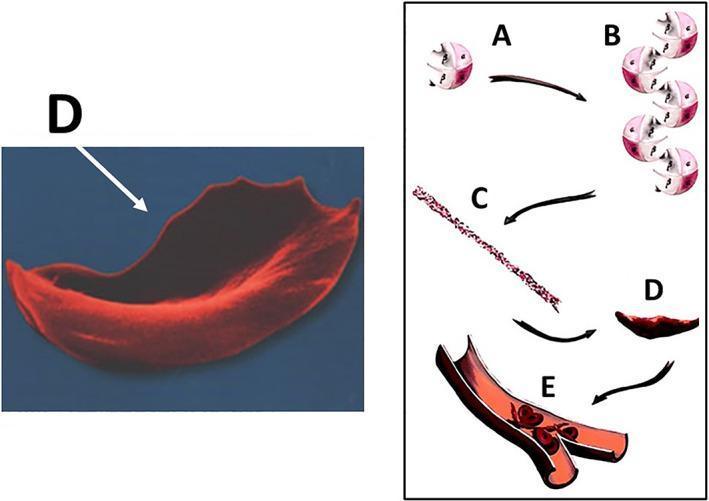
Fig 1: HbS polymerisation cascade leading to sickling and vaso-occlusion
- HbS polymerisation: On deoxygenation, HbS forms long polymeric fibres → distorts RBC into rigid, sickle shape
- Reversibility: Early sickling is reversible on reoxygenation; repeated cycles cause irreversible membrane damage
- Shortened RBC lifespan: Sickled cells survive only ~1/10th normal lifespan (~10–20 days vs. 120 days) → chronic haemolytic anaemia
- Vaso-occlusion: Rigid sickled cells obstruct small blood vessels → stasis → hypoxia → infarction
- Oxidative cascade: HbS auto-oxidation generates reactive oxygen species (ROS), free haem, and iron → endothelial injury, NO depletion, coagulation activation, thrombosis
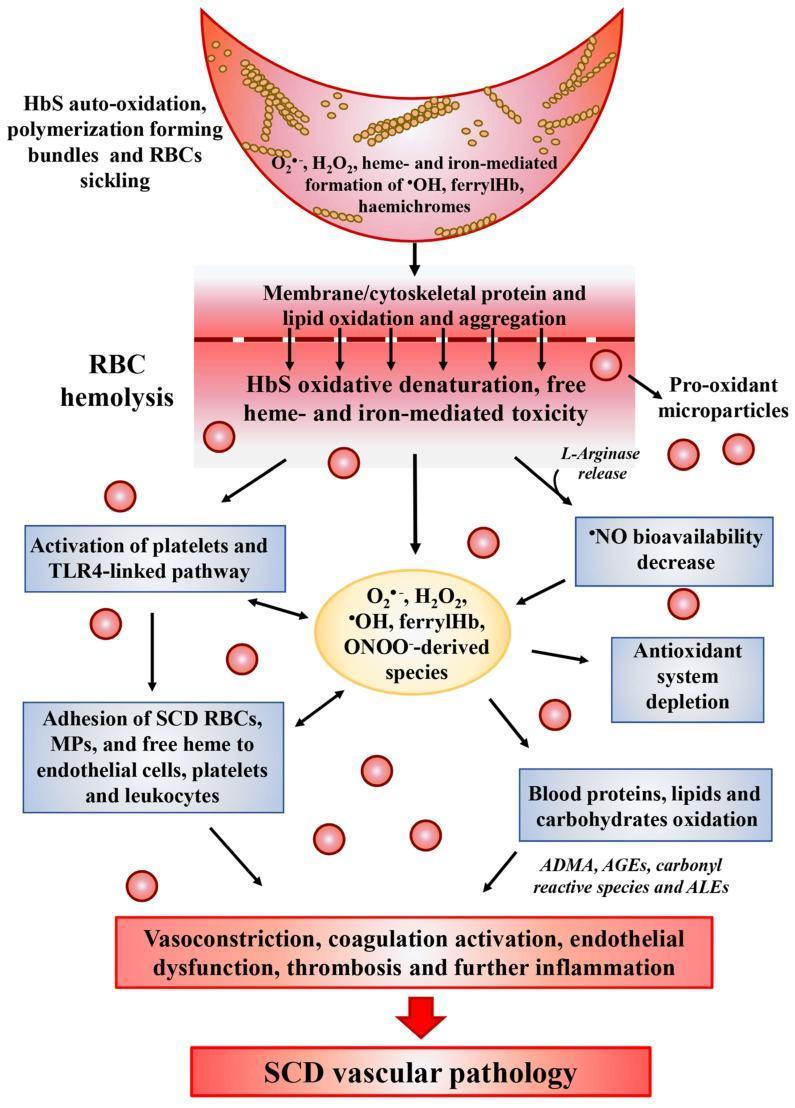
Fig 2: Oxidative stress cascade in SCD — from HbS polymerisation to SCD vascular pathology
Peripheral blood smear shows the classic mixture of sickle cells, target cells, polychromasia, and nucleated RBCs:
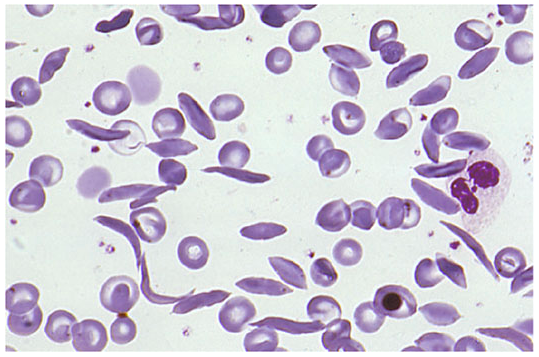
Fig 3: Peripheral blood smear — sickle cells interspersed with normal and target RBCs (Harrison's Principles of Internal Medicine)
4. Precipitating Factors for Sickling Crisis
- Hypoxia (high altitude, anaesthesia, poor ventilation)
- Dehydration
- Cold exposure / temperature extremes
- Infection / fever
- Acidosis
- Stress (physical or emotional)
- Strenuous exercise
5. General Clinical Manifestations (Non-Orthopaedic)
| System | Manifestation |
|---|---|
| Blood | Haemolytic anaemia (Hb 6–9 g/dL), jaundice, cholelithiasis |
| Spleen | Functional asplenia (autosplenectomy by adulthood); acute sequestration in children |
| Lung | Acute chest syndrome (commonest cause of death) |
| CNS | Stroke, silent cerebral infarcts |
| Kidney | Papillary necrosis, nephrotic syndrome, renal failure |
| Eye | Proliferative retinopathy, vitreous haemorrhage |
| Skin | Chronic leg ulcers (medial malleolus) |
| Priapism | Vaso-occlusion in penile vasculature |
Part II — Orthopaedic Complications of Sickle Cell Disease
The orthopaedic manifestations of SCD arise primarily from three mechanisms:
SCD Bone Pathology
│
├── 1. Bone Infarction (vaso-occlusion)
│ ├── Dactylitis (infants)
│ ├── Diaphyseal infarction (metaphysis/epiphysis)
│ └── Osteonecrosis (AVN)
│
├── 2. Marrow Hyperplasia
│ ├── Widened medulla / cortical thinning
│ ├── H-shaped vertebrae
│ └── Osteopenia / pathological fractures
│
└── 3. Osteomyelitis (secondary to infection)
└── Salmonella >> Staphylococcus aureus
Harrison's Principles, Table 386-1; Grainger & Allison's Diagnostic Radiology, p. 1717
Orthopaedic Complication 1: Sickle Cell Dactylitis (Hand-Foot Syndrome)
Definition: Infarction of the bone marrow and cortical bone of the small tubular bones of the hands and feet, resulting in periostitis and soft tissue swelling.
Who gets it: ~50% of infants with SCD between 6 months and 2 years of age; rare after 6 years (when the red marrow of small bones is replaced by fatty marrow, removing the sickling substrate).
Signs & Symptoms:
- Pain, swelling, and warmth of the fingers and/or toes
- Fever (can mimic infection)
- Tender swollen digits
- Lasts 1–3 weeks; resolves without residual damage in most cases
- Asymmetrical shortening of tubular bones is a common long-term sequela
Radiology:
- Early: soft-tissue swelling only
- Later: periosteal elevation, subperiosteal new bone, lytic lesions (bone destruction), irregular cortex
- These changes resolve over months
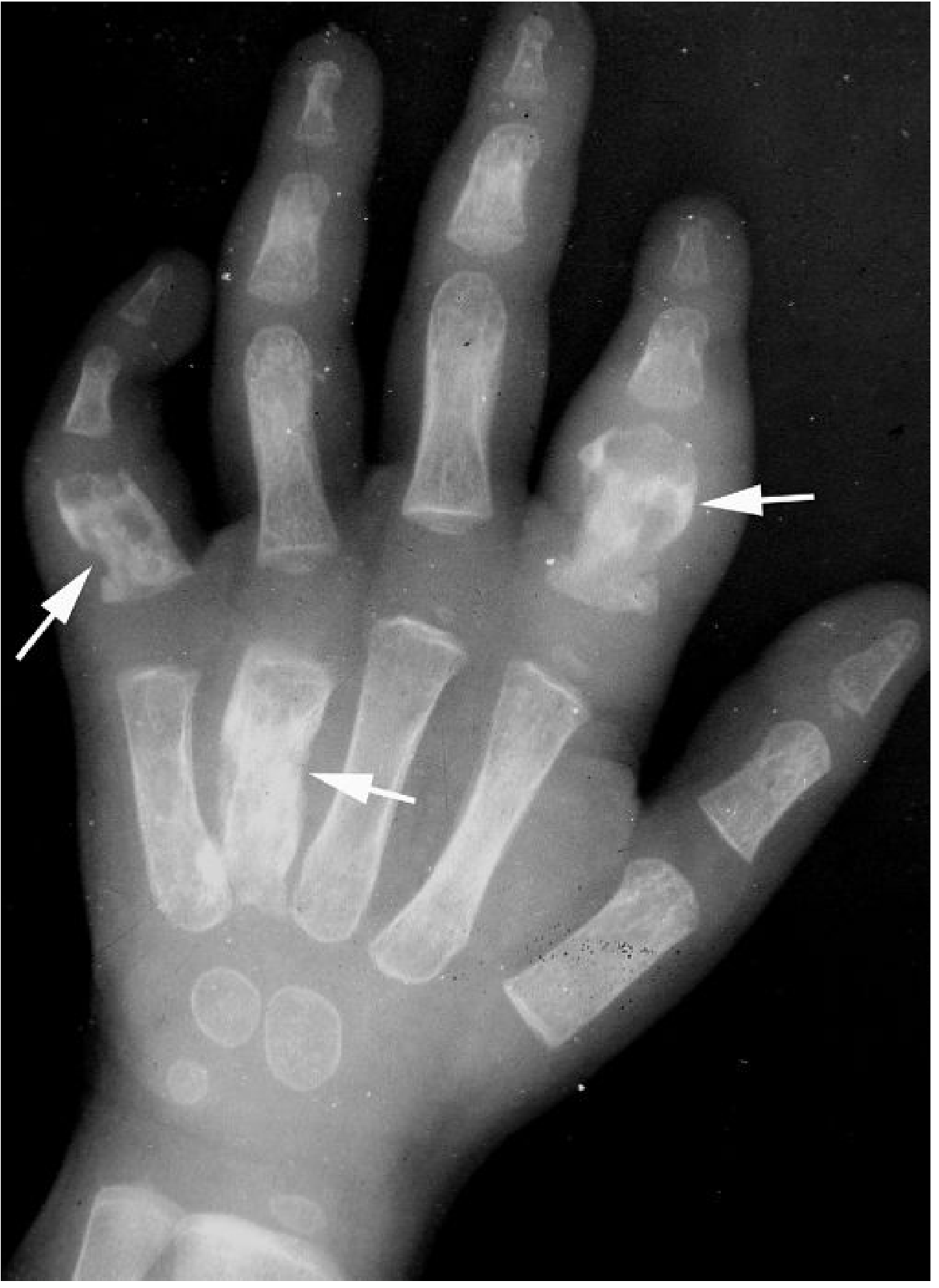
Fig 4: Sickle cell dactylitis — bone destruction at multiple metacarpals and proximal phalanges (white arrows), with soft tissue swelling (Grainger & Allison's, p. 1718)
Treatment: Analgesia, hydration, warmth; antibiotics if infection cannot be excluded. Usually self-limiting.
Orthopaedic Complication 2: Bone Infarction & Vaso-Occlusive Bone Crisis
Pathophysiology: Vaso-occlusion by sickled cells in nutrient vessels → ischaemia → medullary and cortical bone necrosis. Bone infarction is at least 50 times more common than osteomyelitis in SCD.
Sites:
- Children: predominantly diaphysis of long bones
- Adolescents/Adults: metaphyses and epiphyses (→ AVN)
- Ribs/sternum: simulate cardiopulmonary disease
- Vertebrae: central end-plate infarction → "H-shaped" vertebra (~10% of patients)
Signs & Symptoms:
- Severe, deep, constant bone pain (most common reason for hospitalisation)
- Localised tenderness and swelling over affected bone
- Low-grade fever
- Joints may show effusion (periarticular involvement)
- Knees and elbows most commonly affected
Vertebral Involvement — "H-Shaped Vertebra":
Venous thromboembolism in the centre of the vertebral end plate causes focal collapse → central depression → the vertebral body acquires a squared-off "H" or "Lincoln log" shape, almost pathognomonic of SCD:
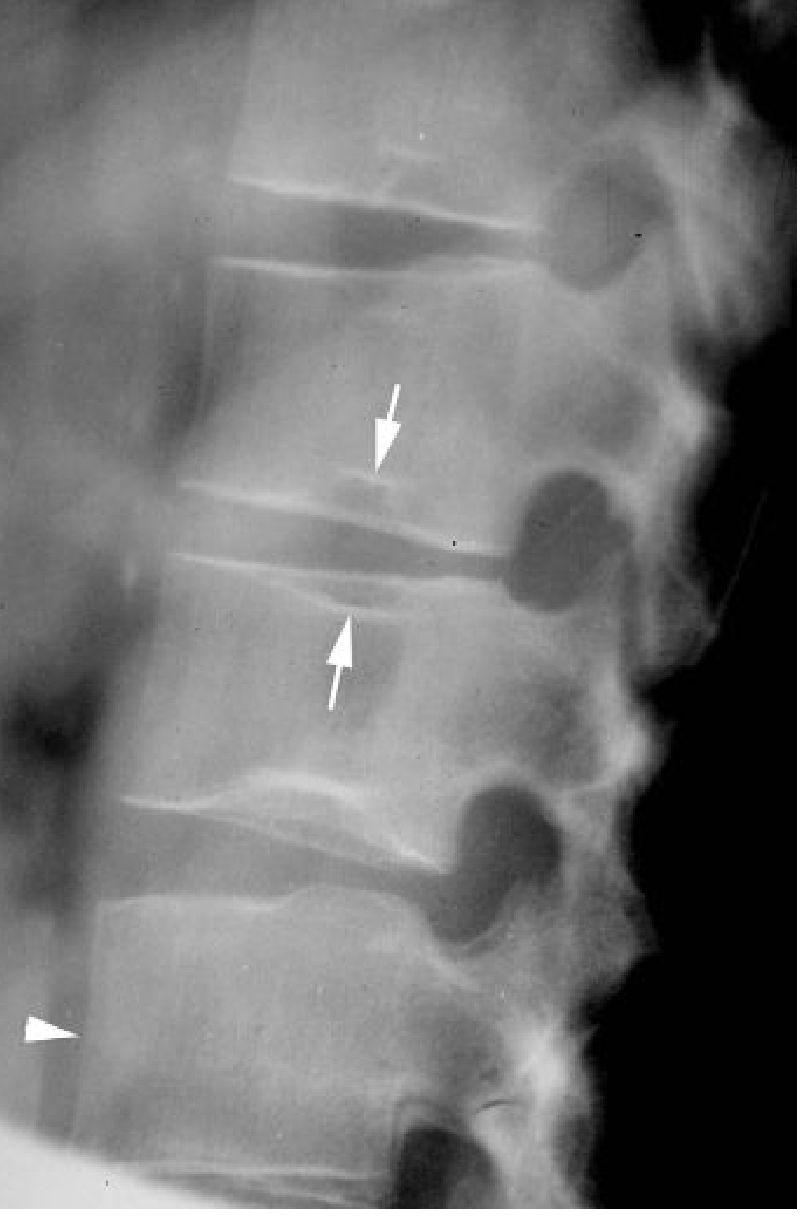
Fig 5: "H-shaped" vertebrae — stepped central depression of multiple lumbar vertebral end plates (white arrows); adjacent "tower" vertebra (arrowhead). (Grainger & Allison's, Fig. 66.15)
Orthopaedic Complication 3: Osteonecrosis / Avascular Necrosis (AVN)
The single most devastating orthopaedic complication of SCD.
Epidemiology:
- ~50% of all SCD patients develop osteonecrosis by age 35
- SCD is the most common cause of osteonecrosis of the femoral head in children
- AVN of the femoral head occurs in ~5% of patients with HbSS
- Paradoxically more common in HbSC (5× more frequent than HbSS, likely due to longer survival)
- Bilateral involvement is frequent
Sites (in order of frequency):
- Femoral head (most common)
- Humeral head
- Distal femur, tibial condyles
- Distal radius
- Vertebral bodies
Pathophysiology: Repeated or sustained vaso-occlusion of the terminal blood supply to the epiphysis → ischaemic death of subchondral bone → subchondral fracture → articular collapse → secondary osteoarthritis.
Signs & Symptoms:
- Insidious onset of pain in groin / hip / shoulder
- Pain worsened by weight bearing and movement
- Reduced range of motion (esp. internal rotation of hip)
- Antalgic gait
- Eventually severe joint destruction with deformity
Investigations:
| Modality | Findings |
|---|---|
| Plain X-ray | Early: normal or subtle sclerosis; Later: patchy radiolucency + sclerosis, subchondral fracture (crescent sign), femoral head collapse, secondary OA |
| MRI (gold standard) | Earliest changes: epiphyseal oedema on STIR (bright signal); T1: low signal fracture line; double-line sign pathognomonic |
| Bone scan | Increased uptake; less specific |
| CT | Confirms subchondral fracture, extent of collapse |
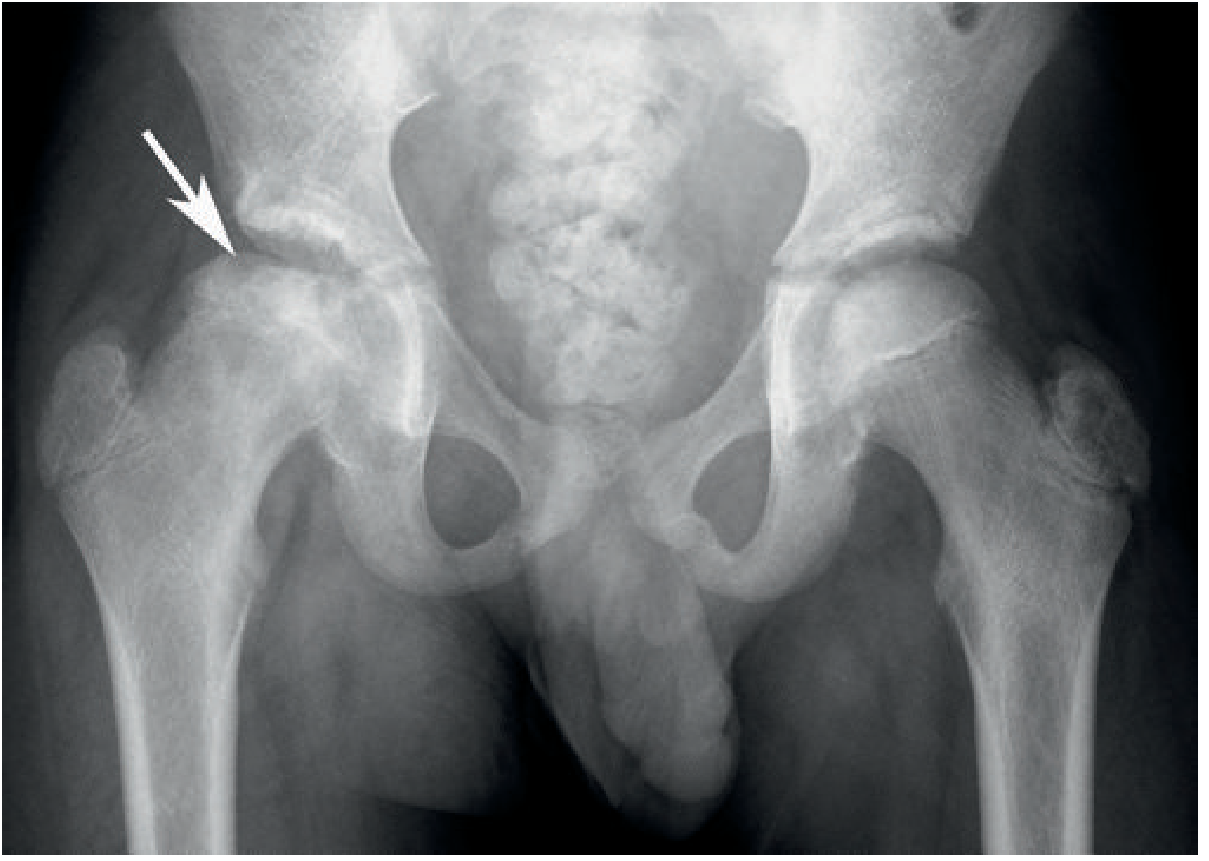
Fig 6: AVN of the right femoral head in SCD — femoral head collapse with patchy lysis and sclerosis (arrow). (Grainger & Allison's, Fig. 66.16)
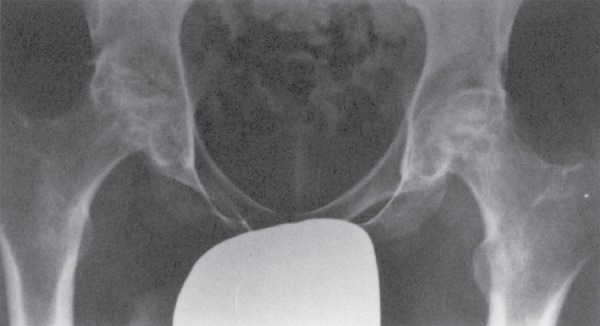
Fig 7: Bilateral femoral head AVN in SCD — advanced bilateral osteonecrosis with femoral head collapse, sclerosis, and secondary OA
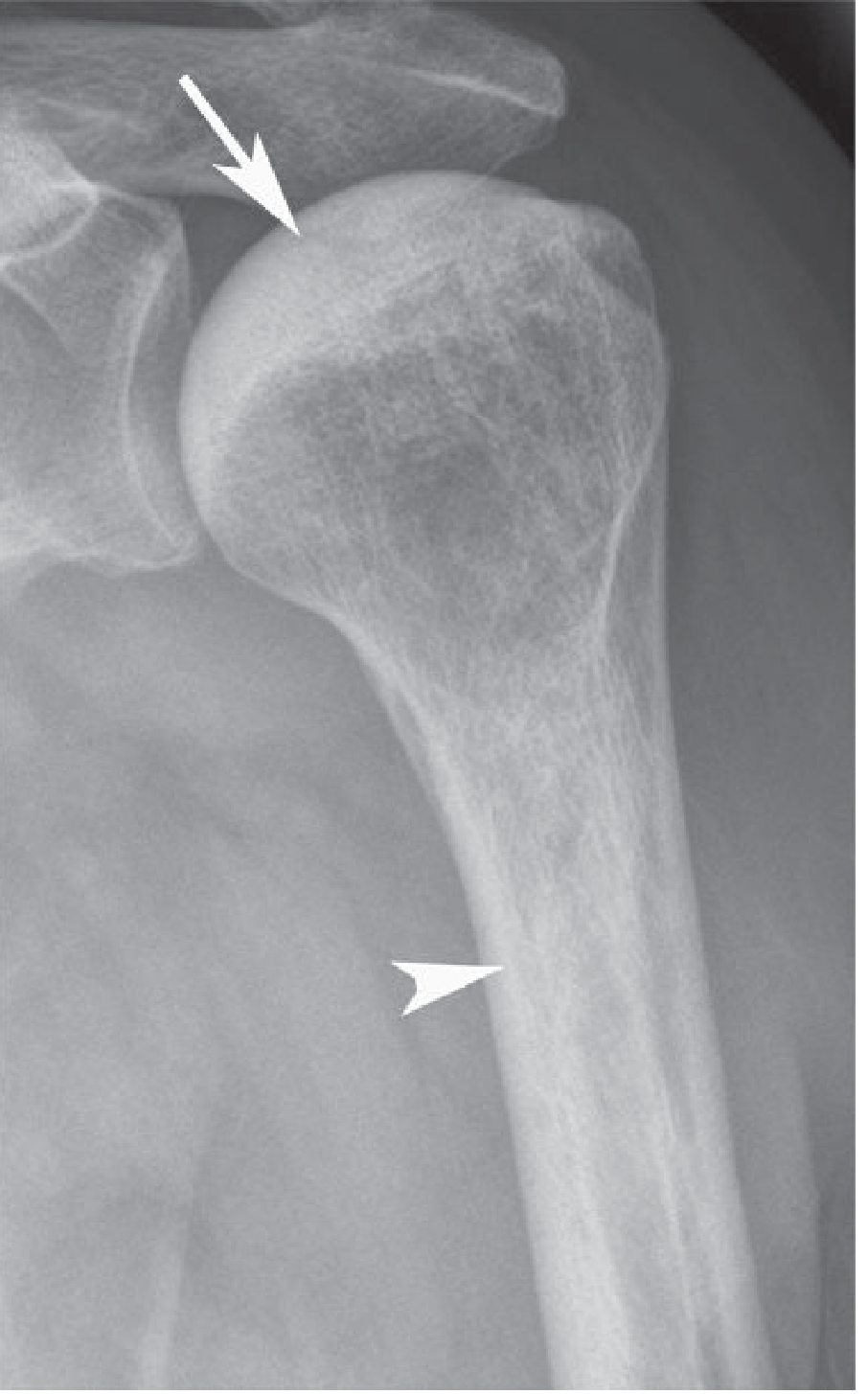
Fig 8: AVN of the humeral head in SCD — epiphyseal sclerosis (arrow) and endosteal sclerosis with narrowing of the medullary cavity (arrowhead). (Grainger & Allison's, Fig. 66.18)
Ficat & Arlet Staging of Femoral Head AVN:
| Stage | Findings |
|---|---|
| I | Normal X-ray; abnormal MRI (bone marrow oedema) |
| II | Sclerosis/cysts; preserved spherical head |
| III | Subchondral fracture (crescent sign); flattening begins |
| IV | Femoral head collapse; secondary OA |
Treatment of AVN:
| Stage | Treatment |
|---|---|
| Early (I–II) | Protected weight bearing; analgesia; physiotherapy; consider core decompression |
| Core decompression | Drilling channels in femoral head to decompress venous hypertension and allow revascularisation; best in Stage I–II |
| Vascularised fibula graft | For Stage II–III in young patients |
| Osteotomy | Rotate necrotic segment away from weight-bearing zone |
| Total Hip Arthroplasty (THA) | Stages III–IV; definitive treatment but technically demanding in SCD due to narrow medullary canal, osteosclerosis, and immunocompromise |
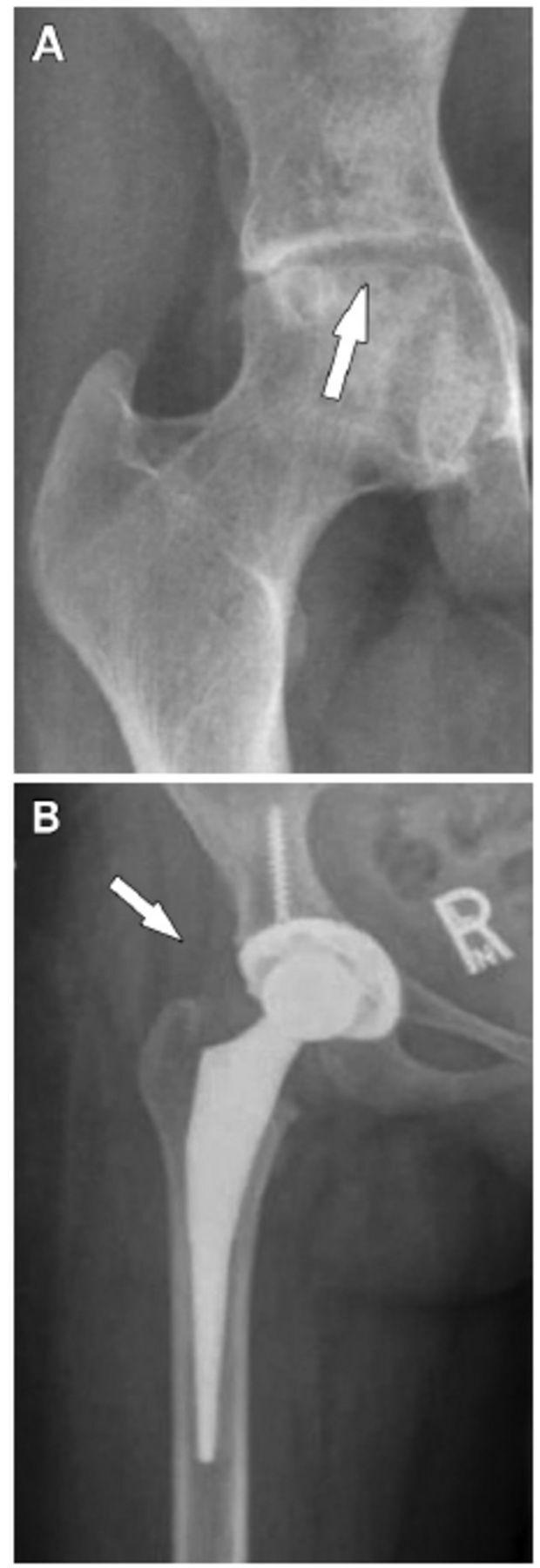
Fig 9: Femoral head AVN (A) treated with total hip arthroplasty (B) — the definitive surgical option for Ficat Stage III–IV
Orthopaedic Complication 4: Osteomyelitis
Unique features in SCD (different from general osteomyelitis):
- Prevalence in SCD: ~18%
- Affects long bone diaphyses (not the metaphysis as in typical childhood osteomyelitis)
- Most common organism: Salmonella spp. (~60–70%) — microinfarcts in bowel allow Salmonella bacteraemia to seed bone
- Second most common: Staphylococcus aureus (~10%)
- Often multifocal
- Mechanism: splenic dysfunction → impaired opsonisation → haematogenous seeding; infarcts provide necrotic substrate for bacterial adhesion
Signs & Symptoms:
- Fever, toxicity, high WBC (more prominent than in bone infarction)
- Localised bone pain, tenderness, swelling
- Often clinically indistinguishable from bone infarction (the "great mimicker")
Key distinction: Osteomyelitis vs. Bone Infarction
| Feature | Osteomyelitis | Bone Infarction |
|---|---|---|
| Fever | High, persistent | Low-grade or absent |
| ESR/CRP | Markedly elevated | Mildly elevated |
| WBC | Markedly elevated | Mildly elevated |
| Response to analgesia | Poor | Improves in 24–48 hours |
| Plain X-ray | Periosteal reaction, lytic destruction | Normal early; sclerosis later |
| MRI | Geographical marrow enhancement post-contrast; cortical defect with abscess | Serpentine enhancement; no cortical defect |
| Bone scan | Hot in all 3 phases | May be cold (photopenic) early |
Treatment:
- Empiric IV antibiotics covering Salmonella + S. aureus: vancomycin + ciprofloxacin (or third-generation cephalosporin)
- Surgical drainage if abscess or sequestrum
- Bone biopsy and culture before antibiotics if possible
Orthopaedic Complication 5: Septic Arthritis
- Prevalence ~7% in SCD
- Haematogenous seeding from bacteraemia (splenic dysfunction) or contiguous osteomyelitis
- Multiple joints may be infected simultaneously
- Common organisms: S. aureus, Streptococcus (Salmonella causes osteomyelitis far more often than septic arthritis)
- Signs & Symptoms: Hot, swollen, tender joint; restricted ROM; fever; joint effusion with high neutrophil count
- Treatment: Urgent joint aspiration/washout + IV antibiotics
Orthopaedic Complication 6: Marrow Hyperplasia & Skeletal Changes
Chronic haemolytic anaemia drives compensatory marrow hyperplasia (red marrow expansion), resulting in:
| Skeletal Change | Description |
|---|---|
| Medullary widening | Expansion of marrow space → cortical thinning |
| Osteopenia | Generalised bone weakness; predisposition to fractures |
| Coarsened trabeculae | Loss of corticomedullary differentiation on X-ray |
| Biconcave vertebrae | Weakened end plates compress; "codfish" appearance |
| H-shaped vertebrae | Central end-plate infarction (see Fig 5) |
| "Bone-within-a-bone" | Periosteal and endosteal cortical thickening from chronic ischaemia |
| Growth disturbance | Epiphyseal growth plate infarction → limb length discrepancy |
Orthopaedic Complication 7: Gouty Arthritis
- Chronic haemolysis → increased nucleic acid turnover → hyperuricaemia
- Acute gouty attacks can occur, particularly in large joints
- Treat as standard gout: colchicine, NSAIDs (use cautiously in SCD), allopurinol for prophylaxis
Part III — Investigations & Evaluation
Haematological & Biochemical
| Test | Finding in SCD |
|---|---|
| Full Blood Count | Hb 6–9 g/dL; MCV normal/low; reticulocytosis (10–20%) |
| Peripheral Blood Smear | Sickle cells, target cells, Howell-Jolly bodies (asplenia), nucleated RBCs, polychromasia |
| Haemoglobin Electrophoresis | Definitive diagnosis: HbS >80% (HbSS); HbA absent in HbSS; HbF variable |
| HPLC (High-Performance Liquid Chromatography) | Gold standard for HbS quantification |
| Reticulocyte count | Elevated (haemolysis marker) |
| LDH / Bilirubin | Elevated (haemolysis) |
| Urine | Haemoglobinuria during crisis |
| ESR / CRP / WBC | Elevated in osteomyelitis > bone infarction |
| Blood cultures | Essential if fever present (septicaemia from asplenia) |
| Uric acid | Elevated → gout risk |
Imaging
| Modality | Role |
|---|---|
| Plain X-ray | First line: H-shaped vertebrae, AVN stages, periosteal reaction, dactylitis, cortical thickening |
| MRI | Gold standard for: early AVN, bone marrow oedema, distinguishing infarction from osteomyelitis, soft tissue involvement |
| Bone scan (Tc-99m) | Sensitive for infarction (may show photopenic "cold" areas early); confirms osteomyelitis (hot in all 3 phases) |
| CT | Assesses cortical destruction, sequestra, extent of femoral head collapse |
| Ultrasound | Joint effusion in septic arthritis |
| DEXA scan | Assess bone mineral density (osteopenia/osteoporosis) |
Neonatal Screening
- Mandatory newborn screening in most countries: heel-prick blood spot HPLC/electrophoresis
- Early diagnosis enables prophylactic penicillin before first sickling crisis
Part IV — Treatment
A. General / Disease-Modifying Treatment
| Treatment | Mechanism / Role |
|---|---|
| Hydroxyurea | Increases HbF production → dilutes HbS → reduces sickling; reduces frequency of crises by ~50%; reduces ACS, stroke, hospitalisations; first-line disease-modifying agent |
| Prophylactic penicillin | Started at 2–3 months of age; prevents pneumococcal sepsis from asplenia; continued until age 5 (some continue lifelong) |
| Folic acid | Supplements demands of chronic haemolysis |
| Vaccinations | Pneumococcal (PCV + PPSV23), meningococcal, Hib, influenza — critical for asplenic patients |
| Blood transfusion | Simple transfusion: acute anaemia, ACS, stroke; Exchange transfusion: acute chest syndrome, stroke, pre-operatively (reduces HbS <30%) |
| Stem cell transplantation | Potentially curative; limited by donor availability; best in children with severe disease and HLA-matched sibling donor |
| Voxelotor | Inhibits HbS polymerisation by increasing Hb oxygen affinity |
| Crizanlizumab | Anti-P-selectin antibody; reduces frequency of VOC |
| L-glutamine | Reduces oxidative damage to RBCs |
| Gene therapy | Emerging; autologous stem cell gene correction (LentiGlobin/Zynteglo) |
B. Orthopaedic-Specific Treatment
Acute Vaso-Occlusive Bone Crisis
- Hydration (IV fluids — reverses sickling, reduces viscosity)
- Analgesia: stepwise — paracetamol → NSAIDs → weak opioids → strong opioids (IV morphine/PCA for severe crisis)
- Warmth of affected extremity
- Oxygen supplementation if hypoxic
- Treat precipitating cause (infection, dehydration)
Osteomyelitis
- IV antibiotics: vancomycin + ciprofloxacin (covers Salmonella and S. aureus)
- Blood cultures and bone biopsy pre-treatment
- Surgical debridement and drainage if abscess, sequestrum, or no response to antibiotics
Avascular Necrosis (AVN)
| Stage | Treatment |
|---|---|
| Precollapse (Ficat I–II) | Non-weight bearing; core decompression (±bone marrow injection); physiotherapy |
| Stage III (early collapse) | Core decompression + vascularised fibular graft; rotational osteotomy in selected young patients |
| Stage IV (established collapse) | Total Hip Arthroplasty (THA) — gold standard; technically challenging (sclerotic/narrow canal); use of cementless stems preferred |
| Humeral head AVN | Shoulder replacement (hemiarthroplasty or TSA) for advanced cases |
Peri-operative SCD precautions (for any orthopaedic surgery):
- Pre-operative exchange transfusion (reduce HbS to <30%)
- Avoid: hypoxia, hypothermia, hypovolaemia, tourniquet use (promotes sickling)
- Adequate post-op hydration and oxygenation
- Early mobilisation
Dactylitis
- Analgesia, hydration, warmth; antibiotics if osteomyelitis cannot be excluded
Summary Table: SCD Orthopaedic Complications at a Glance
| Complication | Key Feature | Most Common Organism | Key Investigation | Definitive Treatment |
|---|---|---|---|---|
| Dactylitis | Age <5; hand/foot swelling | — | X-ray, clinical | Analgesia, hydration |
| Bone infarction | Diaphyseal bone pain; crisis | — | MRI | Analgesia, hydration |
| Osteonecrosis (AVN) | Femoral/humeral head | — | MRI (early); X-ray (late) | Core decompression → THA |
| Osteomyelitis | Diaphyseal; multifocal | Salmonella > S. aureus | MRI, bone biopsy + culture | Vancomycin + ciprofloxacin ± surgery |
| Septic arthritis | Hot, swollen joint | S. aureus, Streptococcus | Joint aspiration, MRI | Washout + IV antibiotics |
| Marrow hyperplasia | H-vertebrae, osteopenia | — | X-ray, MRI, DEXA | Hydroxyurea, calcium/Vit D |
| Gouty arthritis | Large joint attacks | — | Uric acid, joint aspirate | Colchicine, allopurinol |
Key Sources:
- Harrison's Principles of Internal Medicine, 22nd ed. (2025), Ch. 386 — Arthropathies Associated with Haemoglobinopathies
- Grainger & Allison's Diagnostic Radiology, Ch. 66 — Haematological Bone Disease
- Rosen's Emergency Medicine, Ch. 125 — Osteomyelitis in Sickle Cell Disease
- Mulholland and Greenfield's Surgery, 7th ed., Ch. 14 — Sickle Cell Disease
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Ch. 12 — Osteonecrosis
This is a shared conversation. Sign in to Orris to start your own chat.