Adreanl hyperpladia
Reading File
Finding Sources
Finding Sources
Searching PubMed
"adrenal hyperplasia"[MeSH Terms] AND diagnosis
Reading File
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Reading File
I have comprehensive information now. Let me compile the full answer.
Adrenal Hyperplasia
Overview
Adrenal hyperplasia refers to enlargement of the adrenal glands, most commonly due to Congenital Adrenal Hyperplasia (CAH) - a group of autosomal recessive disorders caused by enzyme defects in the cortisol biosynthetic pathway. The fundamental defect is inadequate cortisol synthesis, which triggers compensatory ACTH hypersecretion, causing adrenal gland enlargement (hyperplasia) and accumulation of steroid precursors proximal to the enzymatic block.
Steroidogenesis Pathway
The diagram below shows the complete adrenal and gonadal steroidogenesis pathway, illustrating where each enzyme acts:
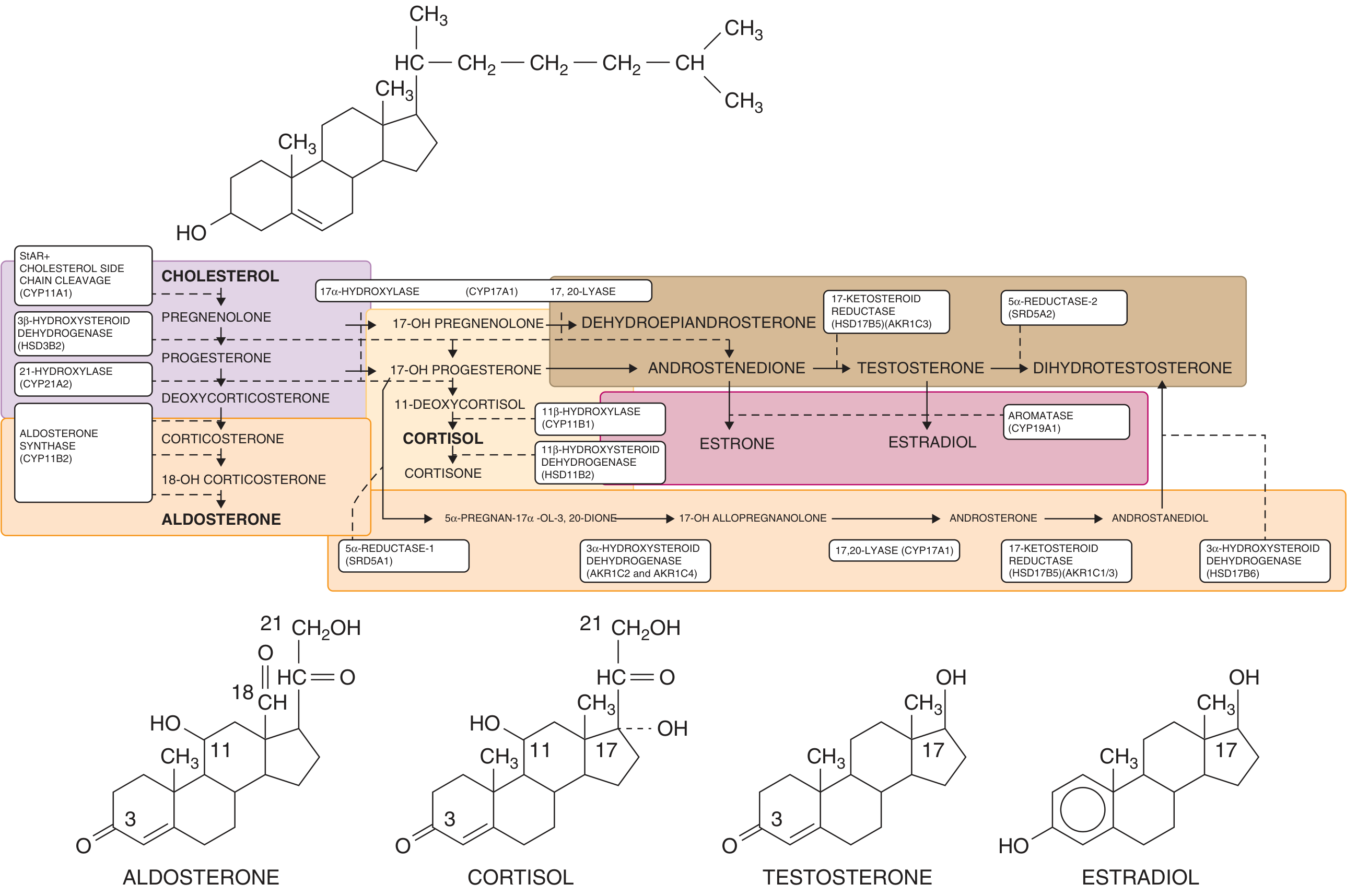
Five enzymes are involved in cortisol biosynthesis. A defect in any one of them can result in CAH:
- Cholesterol side-chain cleavage enzyme (CYP11A1 / StAR)
- 3β-hydroxysteroid dehydrogenase (HSD3B2)
- 17α-hydroxylase (CYP17A1)
- 21-hydroxylase (CYP21A2) - most common
- 11β-hydroxylase (CYP11B1) - second most common
Types of CAH by Enzyme Deficiency
1. 21-Hydroxylase Deficiency (95% of CAH cases)
- Gene: CYP21A2, chromosome 6p21.3 (within the HLA complex); autosomal recessive
- Incidence: 1 in 5,000-15,000 in the US/Europe; highest in Yupik Alaskan Eskimos (1 in 490)
- Over 200 mutations reported; ~10 mutations account for 90-95% of alleles
Clinical subtypes:
| Subtype | Frequency | Features |
|---|---|---|
| Salt-wasting | 75% of classic | Virilization + aldosterone deficiency; risk of adrenal crisis |
| Simple virilizing | 25% of classic | Virilization without salt loss |
| Non-classic (late onset) | Most common autosomal recessive disorder (1:100) | Partial deficiency; hyperandrogenism only |
Clinical features in females:
- Virilization begins in utero at 10 weeks gestation (when external genitalia form)
- Clitoromegaly, labial fusion, urogenital sinus (vagina + urethra sharing a common opening)
- Severity classified by Prader staging (I-V)
- Müllerian structures (uterus, fallopian tubes) are typically NORMAL
Non-classic form in females: Hirsutism, oligo-amenorrhea, male-pattern baldness, polycystic ovaries
Non-classic form in males: Oligospermia, subfertility (reversible with glucocorticoid therapy)
Diagnosis:
- Markedly elevated plasma 17-hydroxyprogesterone (17-OHP) - key diagnostic marker
- Elevated urinary 17-ketosteroids and pregnanetriol
- Pelvic ultrasound confirms müllerian structures (in ambiguous genitalia)
- Neonatal ultrasound may show enlarged, "cerebriform"-appearing adrenal glands
- Neonatal screening (elevated 17-OHP on dried blood spot) - mandatory in all 50 US states and 40+ countries
2. 11β-Hydroxylase Deficiency (~5% of CAH cases)
- Gene: CYP11B1, chromosome 8q (NOT HLA-linked)
- Results in accumulation of 11-deoxycortisol and deoxycorticosterone (DOC)
- Key distinguishing feature: Hypertension (due to mineralocorticoid effect of elevated DOC)
- Classic form: virilization as severe as 21-OH deficiency
- Late-onset form: mild virilization in prepubertal/postpubertal patients
- Diagnosis: Elevated plasma 11-deoxycortisol and 11-deoxycorticosterone
- Treatment: glucocorticoids (same as for 21-OH deficiency)
3. 3β-Hydroxysteroid Dehydrogenase Deficiency (rare)
- Affects early steroidogenesis in both adrenal glands AND gonads
- Impairs synthesis of aldosterone, cortisol, AND sex steroids
- Affected females: mild clitoromegaly + labial fusion + aldosterone/cortisol deficiency
- Affected males: undervirilization (because testosterone synthesis also impaired)
4. Other Forms (rare)
- 17α-hydroxylase deficiency: Hypertension + hypokalemia + sexual infantilism; 46,XX females present with primary amenorrhea; 46,XY males present as phenotypic females
- StAR / cholesterol side-chain cleavage deficiency (lipoid CAH): Most severe form; no steroid hormones produced; lipid accumulation destroys adrenal cortex
Pathophysiology Summary
Enzyme Defect
↓
↓ Cortisol synthesis
↓
Hypothalamus ↑ CRH → Pituitary ↑ ACTH
↓
Adrenal glands become HYPERPLASTIC
Steroid precursors accumulate proximal to the block
↓
Precursors shunted → Androgens (virilization)
Treatment
Glucocorticoid replacement is the cornerstone of treatment for all forms of CAH:
- Suppresses excess ACTH → reduces adrenal hyperplasia and precursor accumulation
- Hydrocortisone (cortisol) is preferred in children (minimizes growth suppression)
- Stress dosing ("sick day rules") is required to prevent adrenal crisis
Mineralocorticoid replacement (fludrocortisone) + salt supplementation:
- Required in salt-wasting 21-OH deficiency
Non-classic CAH: Lower doses of glucocorticoid; treatment only when symptomatic (hirsutism, infertility)
Surgical management (in virilized females):
- Clitoral recession/reduction and vaginoplasty for significant genital ambiguity
- Timing is debated; often done in infancy or deferred to adolescence
Prenatal treatment:
- Maternal dexamethasone (crosses placenta) given early in pregnancy to an at-risk fetus can reduce virilization in affected 46,XX females
- Requires genetic confirmation; ethical considerations involved
Adrenal Medullary Hyperplasia (separate entity)
This is a distinct condition - bilateral enlargement of the adrenal medulla, associated with MEN2 syndromes and predisposing to pheochromocytoma. It is NOT a cortisol synthesis defect.
Key Points Summary
| Feature | 21-OH Deficiency | 11β-OH Deficiency |
|---|---|---|
| Frequency | 95% of CAH | ~5% of CAH |
| Gene | CYP21A2 (chr 6) | CYP11B1 (chr 8) |
| HLA linkage | Yes | No |
| Salt wasting | Yes (75%) | No |
| Hypertension | No | Yes (DOC excess) |
| Virilization | Yes | Yes |
| Elevated marker | 17-OHP | 11-deoxycortisol, DOC |
| Treatment | GC + MC (if salt-wasting) | GC alone |
- Goldman-Cecil Medicine, p. 2515-2516
- Campbell Walsh Wein Urology, pp. 1290-1294
This is a shared conversation. Sign in to Orris to start your own chat.