Hemolytic uremic syndrome
Reading File
Finding Sources
Searching PubMed
"hemolytic uremic syndrome"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Reading File
Finding Sources
Loading Image
Loading Image
Reading File
Reading File
Loading Image
Reading File
I now have extensive, authoritative content from multiple textbooks including Harrison's 2025, Goldman-Cecil, Comprehensive Clinical Nephrology, and others. I have enough to write a thorough, well-structured response.
Hemolytic Uremic Syndrome (HUS)
Overview
Hemolytic uremic syndrome (HUS) is a thrombotic microangiopathy (TMA) characterized by the classic triad of:
- Microangiopathic hemolytic anemia (MAHA)
- Thrombocytopenia
- Acute kidney injury (AKI)
It is the most common cause of acute renal injury requiring renal replacement therapy in children aged 1-5 years, with an annual incidence of ~6.1 cases per 100,000 population.
Classification
HUS is broadly divided into three major forms:
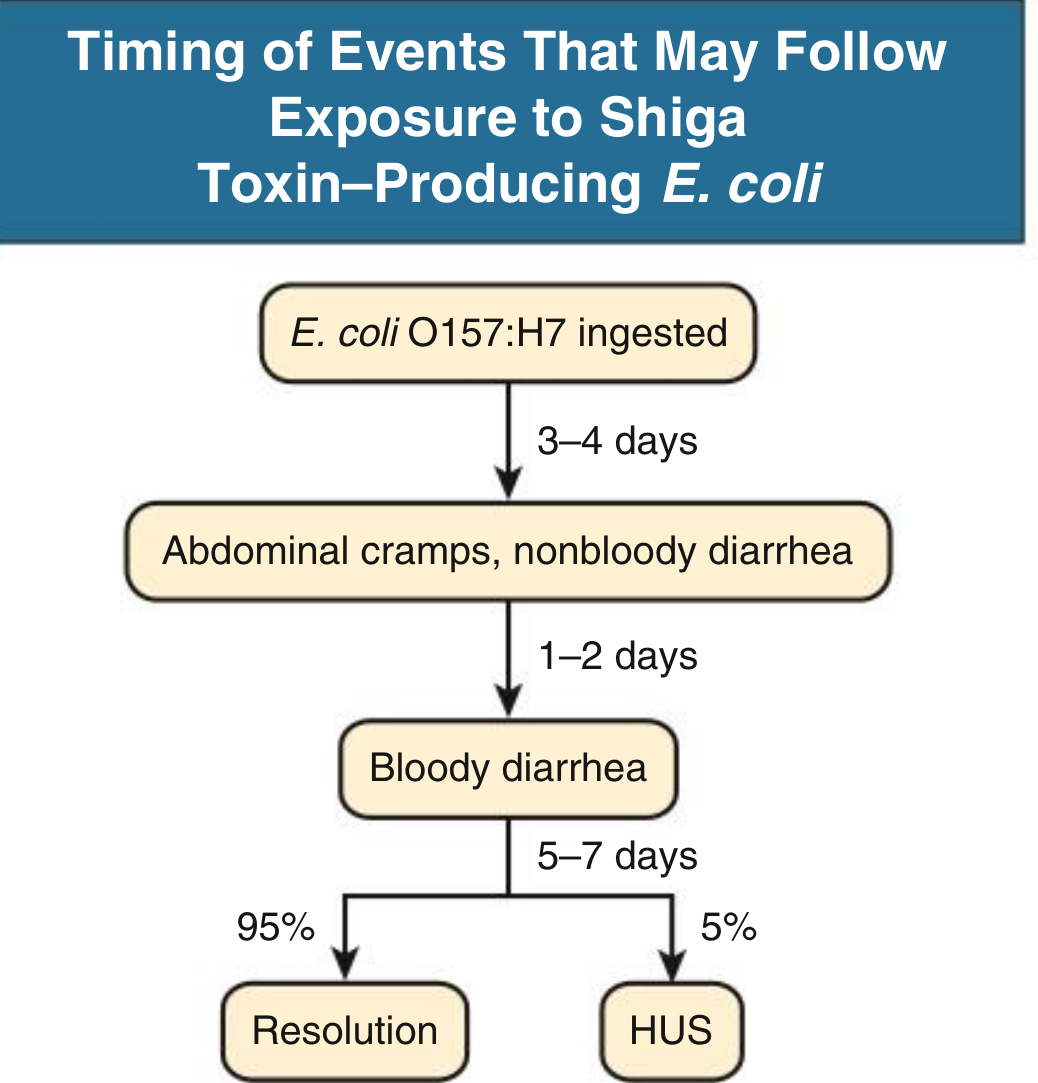
1. Typical (STEC-HUS / D+ HUS) - ~95% of pediatric cases
- Caused by Shiga toxin (Stx)-producing Escherichia coli (STEC), most commonly serotype O157:H7 (also O104:H4, O111, O26, O145)
- Shiga toxins are also produced by Shigella dysenteriae
- Preceded by a bloody diarrheal prodrome in >80% of cases
2. Atypical HUS (aHUS / complement-mediated HUS / D- HUS) - ~5-10%
- Results from dysregulation of the alternative complement pathway
- Genetic mutations or acquired autoantibodies affecting complement regulatory proteins
- Can affect any age; ~67% of aHUS occurs in childhood, but first presentation in adulthood is recognized
3. Secondary HUS
- Caused by drugs (calcineurin inhibitors - tacrolimus, cyclosporine; cytotoxics - mitomycin C, gemcitabine, cisplatin, bleomycin; VEGF inhibitors; proteasome inhibitors)
- Malignancy (gastric, ovarian cancer, leukemia, lymphoma)
- Autoimmune diseases, cancer, pregnancy, bone marrow transplantation
- Streptococcus pneumoniae (produces neuraminidase that exposes Thomsen-Friedereich antigen)
Pathophysiology
Typical (STEC-HUS)
The sequence of events after STEC ingestion:
- After ingestion, Shiga toxins (Stx1, Stx2) enter circulation, often transported on platelets or monocytes
- Toxins bind to globotriaosylceramide (Gb3 / CD77), a glycolipid receptor richly expressed on renal microvasculature endothelium
- Binding induces inflammatory cytokines (IL-8, MCP-1, SDF-1) and chemokine receptors (CXCR4, CXCR7)
- Direct endothelial cell apoptosis/damage
- Release of unusually large von Willebrand factor (vWF) multimers promotes platelet aggregation
- Endothelial damage triggers localized complement activation on the endothelial surface
- Result: platelet thrombi occlude the renal microvasculature; RBCs are sheared as they pass through - forming schistocytes
- The thrombi in STEC-HUS are predominantly fibrin-rich with few platelets (distinguishing from TTP)
Atypical HUS (complement-mediated)
- Defects in the alternative complement pathway regulatory proteins lead to unchecked C3b activation on endothelial surfaces
- Key mutations/deficiencies:
| Gene/Protein | Role | Notes |
|---|---|---|
| Factor H (CFH) | Most common (~50% of aHUS); degrades C3b, prevents C3bBb formation | >70 mutations identified; CFH/CFI mutations carry >50% mortality or ESKD |
| Factor I (CFI) | Proteolytically cleaves C3b and C4b | |
| Membrane Cofactor Protein (MCP/CD46) | Cofactor for Factor I | |
| Factor B | Component of C3 convertase (C3bBb) | |
| C3 | Direct mutations described | |
| Thrombomodulin | Cofactor in complement regulation | |
| CFHR1, CFHR3, CFHR5 | Related complement regulatory proteins |
- DEAP-HUS: Deficiency of CFHR plasma proteins and autoantibody-positive - ~6% of patients have autoantibodies against CFH ("autoimmune HUS"); almost all diagnosed before age 16
- Trigger events (URI, oral contraceptives, pregnancy) can precipitate HUS in genetically susceptible individuals
- Oral contraceptives trigger HUS in ~8% of CFH-mutant and ~20% of CFI-mutant patients
Clinical Features
| Feature | Typical (STEC) HUS | Atypical HUS |
|---|---|---|
| Age | Children <5 years predominantly; adults susceptible | Any age; first presentation possible in adulthood |
| Prodrome | Bloody diarrhea, abdominal cramps, vomiting | Often URI; no diarrhea |
| Fever | Usually absent | Variable |
| Renal failure | Universal; hallmark | Universal; often severe and progressive |
| Neurological | <50% (dysphasia, hyperreflexia, encephalopathy, seizures, cerebral infarction - esp. adults) | Present but variable |
| Hypertension | Common | Common |
| Recurrence | Rare after recovery | >75% with CFH/CFI mutations |
| Skin | Petechiae, retiform purpura (uncommon) | Uncommon |
Laboratory Findings
- Peripheral blood smear: schistocytes (fragmented RBCs) - diagnostic hallmark
- Anemia: normocytic/normochromic, often severe (may require RBC transfusions in up to 80%)
- Thrombocytopenia: platelets typically <100,000/µL
- Elevated LDH: marker of hemolysis
- Decreased haptoglobin
- Elevated unconjugated bilirubin
- Negative direct Coombs test (non-immune hemolysis)
- Elevated serum creatinine: subacute worsening pattern
- Urinalysis: hematuria, proteinuria, granular/hyaline casts
- Normal coagulation (PT, aPTT usually normal - distinguishes from DIC)
- Complement studies (in aHUS): low C3, normal C4 (alternative pathway activation); check factor H, I levels; screen for CFH autoantibodies
- ADAMTS13 activity: near-normal in HUS (vs. severely reduced in TTP)
Histopathology
The hallmark is thrombotic microangiopathy (TMA) of the renal vasculature:
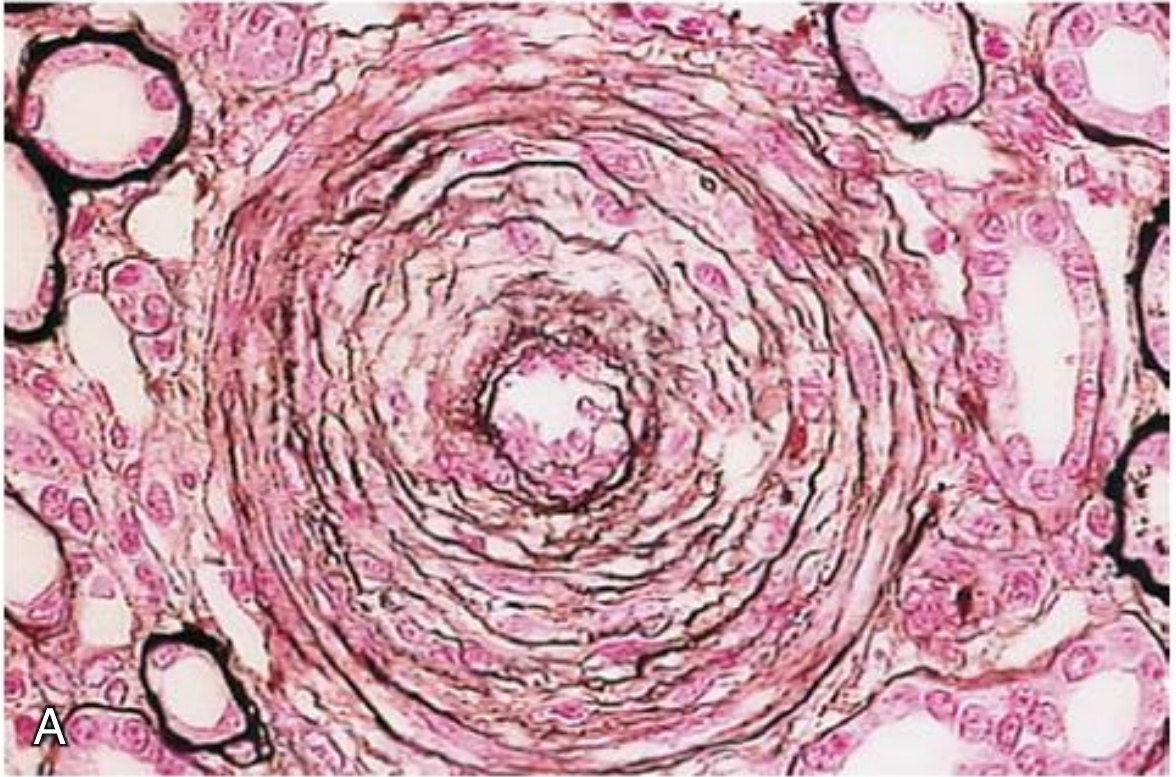
Interlobular artery in severe vascular HUS - showing myointimal proliferation, reduplication of the elastic lamina, and near-complete luminal occlusion with thrombotic material:
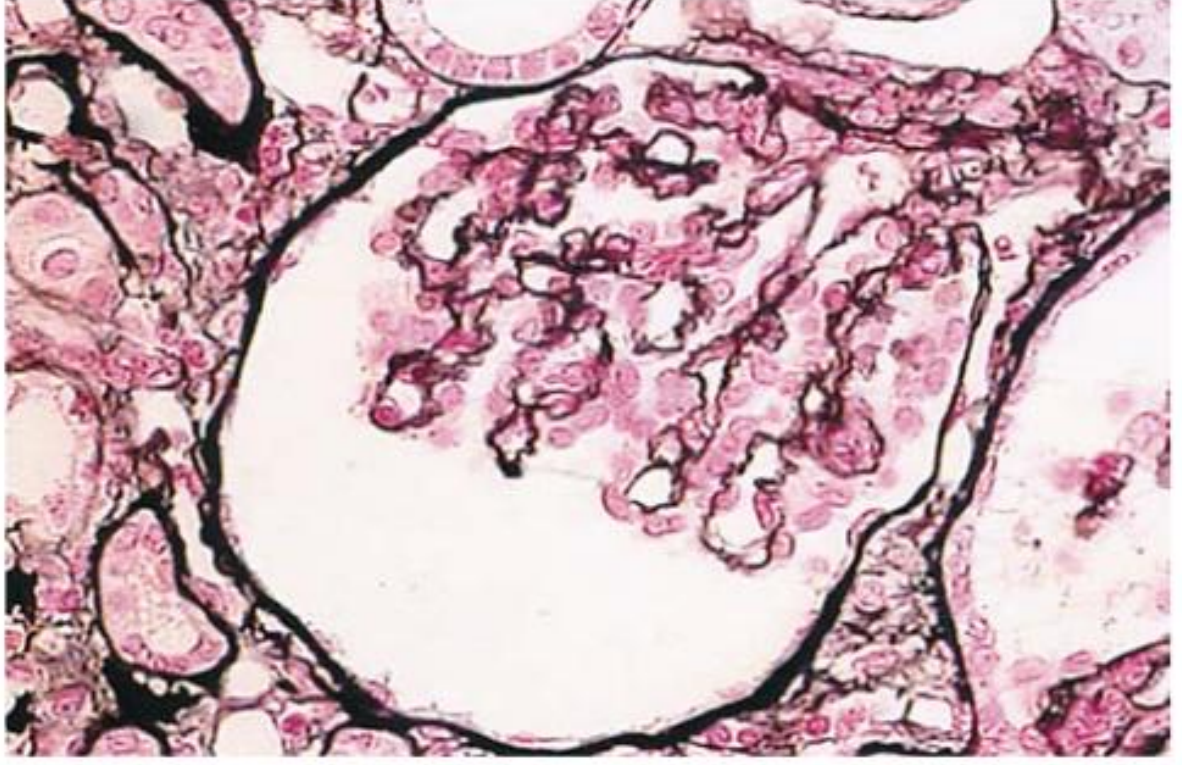
Glomerulus in atypical HUS - showing severe ischemic changes, shrinkage of the glomerular tuft, and marked thickening and wrinkling of capillary walls:
Additional findings:
- Patchy or diffuse renal cortical necrosis in up to 40% of severe cases
- Arterioles and small arteries show intimal proliferation with luminal stenosis
- Thrombi in STEC-HUS are fibrin-predominant; in aHUS, platelet-rich thrombi may predominate
Differential Diagnosis: HUS vs. TTP
| Feature | HUS | TTP |
|---|---|---|
| Age | Children predominantly (typical) | Adults (peak: 3rd decade, more in females) |
| Renal failure | Predominant | Mild or absent |
| Neurological involvement | <50% in typical; variable in aHUS | Prominent (historically) |
| Fever | Usually absent | Present (classically) |
| ADAMTS13 | Normal or mildly reduced | Severely reduced (<10%) |
| Preceding diarrhea | Yes (typical HUS) | No |
| Plasma exchange | Limited benefit (typical); variable in aHUS | First-line therapy |
Treatment
Typical STEC-HUS - Supportive management is the cornerstone
- IV isotonic volume expansion early (even before culture results), to limit AKI severity and reduce need for dialysis
- RBC transfusions - up to 80% of patients require them
- Renal replacement therapy (dialysis) for severe AKI
- Bowel rest / parenteral nutrition for the associated enterohemorrhagic colitis
- Blood pressure control; long-term RAS blockade if CKD develops
- Avoid heparin and anticoagulants (increases bleeding risk), antimotility agents (prolong toxin exposure), antiplatelets
Antibiotics: Generally contraindicated in STEC-HUS in children - antibiotic therapy may increase HUS risk up to 17-fold (due to acute release of large amounts of preformed toxin from lysed bacteria; quinolones and trimethoprim also induce Stx gene expression). Exception: azithromycin may reduce bacterial shedding; antibiotics indicated for Shigella bacteremia.
- Plasma exchange: Unproven in typical STEC-HUS; may be considered in severe adult cases with AKI and CNS involvement
- Eculizumab in STEC-HUS: Controversial - case reports of benefit exist, but a trial during the 2011 German O104:H4 outbreak showed no clear benefit over plasma exchange alone
Atypical (Complement-mediated) HUS
- Eculizumab (anti-C5 humanized monoclonal antibody): FDA-approved first-line therapy for aHUS - blocks terminal complement activation; effective for CFH/CFI mutations
- Ravulizumab: Engineered from eculizumab, extended half-life, FDA-approved for aHUS; both require mandatory meningococcal vaccination before initiation
- Plasma exchange/infusion: May temporize by replacing deficient complement regulatory proteins (esp. for CFH deficiency), but far less effective than in TTP
- Immunosuppressives (prednisone, rituximab, MMF): Used in autoimmune aHUS (anti-CFH antibodies); rituximab beneficial for anti-ADAMTS13-deficient TTP and sometimes for chemotherapy-induced HUS
- Eculizumab discontinuation: Current evidence (2025 meta-analysis, PMID 40795230) is being evaluated - some patients may safely discontinue under monitoring
Secondary HUS
- Remove causative agent (stop offending drug, lower calcineurin inhibitor dose)
- Plasma exchange, immunosuppression, rituximab have been used with variable success
- Eculizumab considered for complement-mediated secondary forms
- Prognosis is generally poor (most chemotherapy-induced HUS patients die within months)
Prognosis
| Form | Outcome |
|---|---|
| Typical STEC-HUS | Mortality reduced to 5-15% with modern supportive care (from ~50% historically); ~50% have minor to major residual renal impairment; recurrence rate 0-10% post-transplant |
| aHUS (CFH/CFI mutations) | >50% mortality or ESKD without treatment; recurrence in >75% with CFH/CFI mutations; pre-eculizumab era: poor |
| aHUS (MCP mutation) | Better prognosis; MCP is a membrane protein not replaced by plasma, but the kidney graft expresses MCP |
| Secondary HUS | Generally poor; depends on underlying cause |
Recent Evidence (2024-2026)
- A 2025 systematic review and meta-analysis examined extrarenal manifestations of aHUS - confirming that cardiac, neurological, GI, and pulmonary involvement are more common than historically recognized (PMID 39676096)
- A 2025 systematic review assessed eculizumab discontinuation in aHUS - relevant for clinical decision-making on treatment duration (PMID 40795230)
- A 2025 meta-analysis reviewed kidney and pregnancy outcomes in pregnancy-associated aHUS, a high-risk subset (PMID 39889176)
Sources: Andrews' Diseases of the Skin, p. 3325 | Goldman-Cecil Medicine, p. 1199 | Harrison's Principles of Internal Medicine 22E (2025), Chaps. 80 & 329 | Comprehensive Clinical Nephrology 7th Ed., p. 424 | Henry's Clinical Diagnosis, p. 711 | NKF Primer on Kidney Diseases 8e | Brenner & Rector's The Kidney
Hus vs TTP
Finding Sources
Searching PubMed
"thrombotic thrombocytopenic purpura" AND "hemolytic uremic syndrome" AND ADAMTS13
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
I have comprehensive content from multiple authoritative sources. Now I'll compile the full HUS vs TTP comparison.
HUS vs TTP: A Detailed Comparison
Both are thrombotic microangiopathies (TMAs) sharing the core features of microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and organ ischemia. However, they are now understood as distinct entities with different causes, dominant organ involvement, and treatments.
Pathophysiology - The Fundamental Difference
| TTP | HUS (Typical / STEC) | Atypical HUS (aHUS) | |
|---|---|---|---|
| Core defect | Severe deficiency of ADAMTS13 (<10% activity) | Endothelial damage from Shiga toxin (Stx1/Stx2) | Dysregulation of the alternative complement pathway |
| Mechanism | ADAMTS13 normally cleaves ultra-large vWF (ULvWF) multimers. Without it, ULvWF persists, binds and activates platelets → platelet-rich thrombi in microvasculature | Stx binds Gb3 receptor on renal endothelium → apoptosis, cytokine release, complement activation → platelet thrombi; Stx also inhibits endothelial ADAMTS13 production | Mutations/autoantibodies in complement regulatory proteins (Factor H, I, MCP, C3, Factor B, thrombomodulin) → unchecked C3b activation on endothelial surfaces → endothelial damage + intravascular thrombosis |
| Thrombus composition | Platelet-rich, vWF-rich | Fibrin-predominant, few platelets | Platelet-rich (similar to TTP) |
| Cause - Hereditary | Congenital TTP (cTTP / Upshaw-Schulman syndrome): homozygous/compound heterozygous ADAMTS13 mutations | - | Mutations in CFH, CFI, MCP (CD46), C3, CFB, CFHR1/3/5, thrombomodulin |
| Cause - Acquired | Acquired/immune TTP (iTTP): anti-ADAMTS13 autoantibodies (most common adult form) | STEC infection (O157:H7, O104:H4, etc.) | Anti-CFH autoantibodies (DEAP-HUS, ~6%) |
Demographics
| TTP | STEC-HUS | aHUS | |
|---|---|---|---|
| Predominant age | Adults (peak 3rd decade); cTTP in children | Children <5 years (also adults) | Any age; ~67% childhood onset, but adult presentation recognized |
| Sex | More common in females | Equal | Equal |
| Incidence | 2.88 per 10^6/year (adults); 0.09 per 10^6/year (children) | ~6-8 per 100,000/year (STEC infection) | Rare |
| Trigger | Pregnancy, autoimmune disease, HIV, drugs, idiopathic | STEC-contaminated food/water | Infection (URI), pregnancy, OCP use, idiopathic |
Clinical Features
| Feature | TTP | STEC-HUS | aHUS |
|---|---|---|---|
| Classic pentad | MAHA + thrombocytopenia + neurological + fever + renal (seen in ~88-98%) | MAHA + thrombocytopenia + renal failure ± diarrhea prodrome | MAHA + thrombocytopenia + renal failure (often severe/progressive) |
| Fever | Present (classically) | Usually absent | Variable |
| Renal failure | Mild (can occur in up to 27%) | Dominant feature - universal | Dominant, often severe, progressive to ESKD |
| Neurological | Prominent: headache, confusion, focal deficits, seizures, coma | Present in <50%; more common in adults with severe disease | Present in 10-30% |
| Diarrhea prodrome | Absent | Bloody diarrhea in >80% (3-7 days before HUS onset) | Absent (URI more typical trigger) |
| Cardiac involvement | Myocardial infarction, arrhythmias, cardiogenic shock | Uncommon | Uncommon in typical; systemic in DEAP-HUS |
| Skin | Petechiae (due to thrombocytopenia) | Petechiae, retiform purpura (uncommon) | Uncommon |
| Recurrence | Chronic relapsing disorder; most relapses in 1st-2nd year but can occur decades later | Rare after recovery | >75% with CFH/CFI mutations |
Timing of STEC-HUS onset (for reference):
Laboratory Findings - Key Differences
| Lab | TTP | HUS |
|---|---|---|
| ADAMTS13 activity | <10% - pathognomonic for TTP | Normal or mildly reduced |
| Anti-ADAMTS13 antibodies | Present in iTTP | Absent |
| Platelet count | Typically <30 × 10⁹/L (severe) | Typically >30 × 10⁹/L |
| Serum creatinine | Mildly elevated or normal | Markedly elevated |
| Complement (C3, C4) | Normal | C3 low, C4 normal in aHUS (alternative pathway activation) |
| Factor H level / anti-CFH Ab | Normal | Abnormal in aHUS |
| Schistocytes on smear | Present | Present |
| LDH | Markedly elevated | Markedly elevated |
| Haptoglobin | Decreased/absent | Decreased/absent |
| Coombs test | Negative | Negative |
| Coagulation (PT/aPTT) | Normal | Normal (distinguishes from DIC) |
| Urinalysis | Mild hematuria/proteinuria | Marked hematuria, proteinuria, casts |
PLASMIC Score (Clinical TTP Probability Tool)
Used when ADAMTS13 results pending - a score ≥5/7 predicts severe ADAMTS13 deficiency (TTP):
| Variable | Points |
|---|---|
| Platelet count <30 × 10⁹/L | +1 |
| Hemolysis (reticulocytosis or bilirubin >2 mg/dL) | +1 |
| Absence of active cancer | +1 |
| Absence of solid-organ or stem cell transplant | +1 |
| MCV <9.0 fL (no macrocytosis) | +1 |
| INR <1.5 | +1 |
| Creatinine <2.0 mg/dL | +1 |
Treatment - Side by Side
| Treatment | iTTP | cTTP | STEC-HUS | aHUS |
|---|---|---|---|---|
| Plasma exchange (PLEX) | First-line, urgent - removes anti-ADAMTS13 antibodies + replaces ADAMTS13; PLEX superior to plasma infusion (78% vs 49% 6-month survival) | Plasma infusion (PI) usually sufficient | Not recommended (no proven benefit; may consider in severe adults with CNS involvement) | May temporize (replaces CFH) but inferior to eculizumab |
| Corticosteroids | Yes - in combination with PLEX; reduces number of PLEX sessions | - | Not helpful | In autoimmune aHUS (anti-CFH Ab) |
| Rituximab (anti-CD20) | Yes - frontline with PLEX for refractory/relapsing iTTP; depletes anti-ADAMTS13 antibody-producing B cells | - | Not indicated | In autoimmune aHUS (anti-CFH Ab) |
| Caplacizumab | Approved adjunct - anti-vWF nanobody blocking vWF-platelet interaction at A1 domain; hastens platelet recovery, reduces death/recurrence/thromboembolism (TITAN + HERCULES trials); used with PLEX + immunosuppression | - | Not indicated | Not indicated |
| Eculizumab / Ravulizumab | Not indicated (unless complement-mediated overlap) | - | Controversial in STEC-HUS (no proven benefit in German outbreak RCT) | First-line - anti-C5 antibody blocks terminal complement; FDA-approved for aHUS; meningococcal vaccination required |
| Recombinant ADAMTS13 | Under investigation (promising in phase 1 cTTP trial) | Emerging therapy | Not applicable | Not applicable |
| Antibiotics | Not applicable | - | Generally contraindicated in children (risk of HUS ↑17-fold); azithromycin may reduce shedding in adults | Not applicable |
| Supportive dialysis | Rarely needed | - | Cornerstone of management (up to 60-80% may need RRT) | Often needed; aggressive BP control |
| Bortezomib | Rescue in rituximab-refractory iTTP | - | - | - |
Summary Comparison Table
| Feature | TTP | STEC-HUS | aHUS |
|---|---|---|---|
| Pathogenesis | ADAMTS13 deficiency (congenital or autoimmune) | Shiga toxin - endothelial damage + complement | Alternative complement dysregulation (genetic/autoimmune) |
| Key patient | Adult female | Child <5 years | Any age |
| Diarrhea prodrome | No | Yes (bloody) | No |
| Dominant organ | Brain | Kidney | Kidney |
| ADAMTS13 | <10% | Normal | Normal |
| Complement | Normal | Normal (may activate secondary) | C3 low, C4 normal |
| Platelet count | Often <30 × 10⁹/L | Often >30 × 10⁹/L | Often >30 × 10⁹/L |
| Mortality untreated | ~90% | 5-15% with support | >50% (CFH/CFI mutations) |
| First-line Rx | Plasma exchange + steroids + caplacizumab ± rituximab | Supportive care | Eculizumab or ravulizumab |
| Recurrence | Chronic (lifelong risk) | Rare | High (>75% in CFH/CFI mutations) |
Key clinical pearl: ADAMTS13 activity is the single most important test to differentiate TTP from HUS. A level <10% is pathognomonic for TTP. Because the test takes time, a PLASMIC score ≥5 should prompt immediate plasma exchange while awaiting results - as untreated TTP carries ~90% mortality. HUS, by contrast, does not benefit from plasma exchange in the typical form and requires eculizumab specifically for the complement-mediated variant.
Sources: Brenner & Rector's The Kidney | Harrison's Principles of Internal Medicine 22E (2025) | Goldman-Cecil Medicine | Comprehensive Clinical Nephrology 7th Ed. | Washington Manual of Medical Therapeutics
Infarct classification as per duration
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Reading File
Finding Sources
Finding Sources
Reading File
Loading Image
Loading Image
I now have all the content needed - the complete morphological table from Robbins, both gross and microscopic images. Here is the comprehensive answer:
Infarct Classification by Duration (Age)
This refers primarily to myocardial infarction (MI) - the most systematically studied infarct in pathology. The classification is based on the sequential gross and microscopic changes that occur as the infarcted tissue progresses from necrosis through inflammation to scarring.
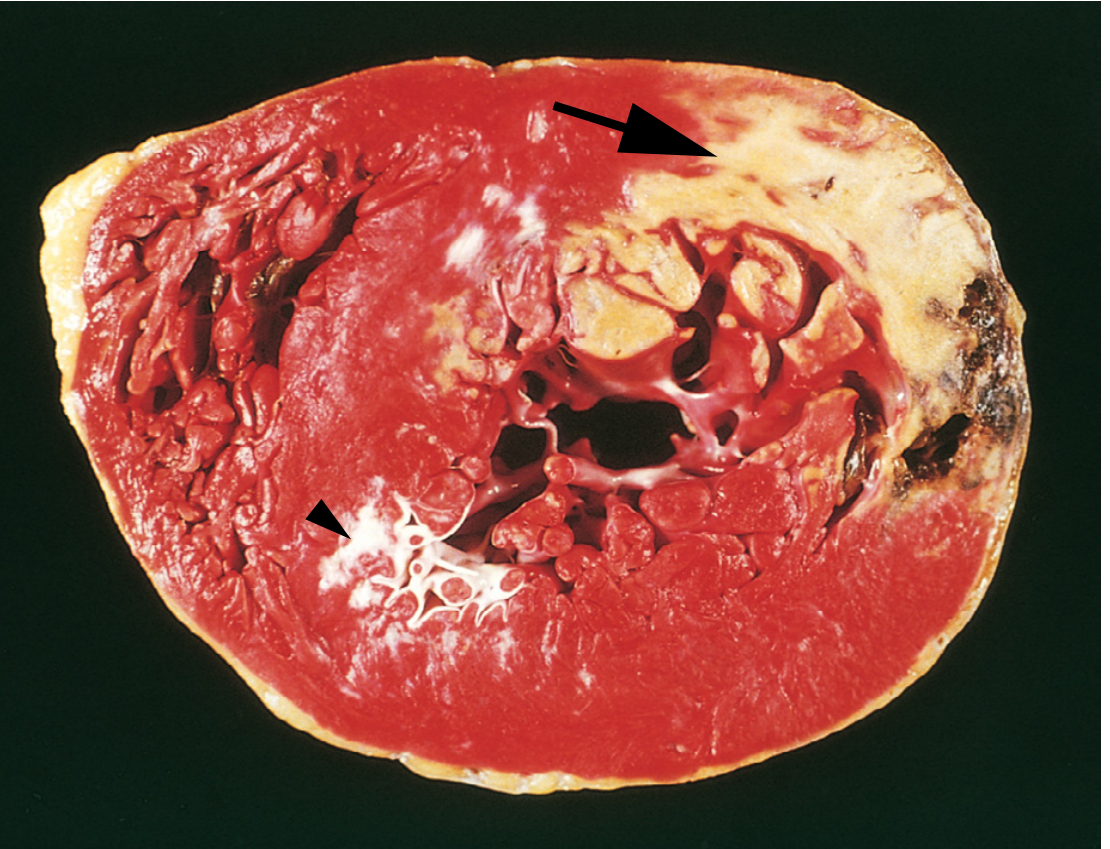
Gross: Acute MI - TTC Staining
Triphenyltetrazolium chloride (TTC) stain is used at autopsy to identify early infarcts that are not yet visible to the naked eye. Viable myocardium stains brick-red (contains LDH enzyme); necrotic myocardium is unstained/pale (enzyme has leaked out). Old scars appear white and glistening.
Complete Morphological Timeline (Robbins Classification)
| Time Frame | Stage | Gross Features | Light Microscopy | EM Findings |
|---|---|---|---|---|
| 0 - 30 min | Reversible injury | None | None | Relaxation of myofibrils; glycogen loss; mitochondrial swelling |
| 30 min - 4 hrs | Early irreversible | None | Usually none; wavy fiber change at border (dead fibers stretched/buckled by adjacent viable myocytes) | Sarcolemmal disruption; mitochondrial amorphous densities |
| 4 - 12 hrs | Early irreversible | Occasionally dark mottling | Onset of coagulative necrosis; edema; hemorrhage | - |
| 12 - 24 hrs | Acute | Dark mottling | Ongoing coagulative necrosis; pyknosis of nuclei; hypereosinophilic myocytes; marginal contraction band necrosis; early neutrophilic infiltrate | - |
| 1 - 3 days | Acute | Mottling with yellow-tan center | Coagulative necrosis with loss of nuclei and striations; increased neutrophils (peak acute inflammation) | - |
| 3 - 7 days | Subacute | Hyperemic border; central yellow-tan softening | Disintegration of dead myofibers; dying neutrophils; early macrophage phagocytosis at borders | - |
| 7 - 10 days | Subacute | Maximally yellow-tan and soft; depressed red-tan margins | Well-developed phagocytosis; early granulation tissue at margins | - |
| 10 - 14 days | Subacute/Healing | Red-gray depressed borders | Well-established granulation tissue with new blood vessels + collagen deposition | - |
| 2 - 8 weeks | Healing | Gray-white scar progressing from border to core | Increased collagen deposition; decreased cellularity | - |
| >2 months | Healed (Old) | Scarring complete | Dense collagenous scar - identical whether 8 weeks or 10 years old |
Histological Stages Illustrated
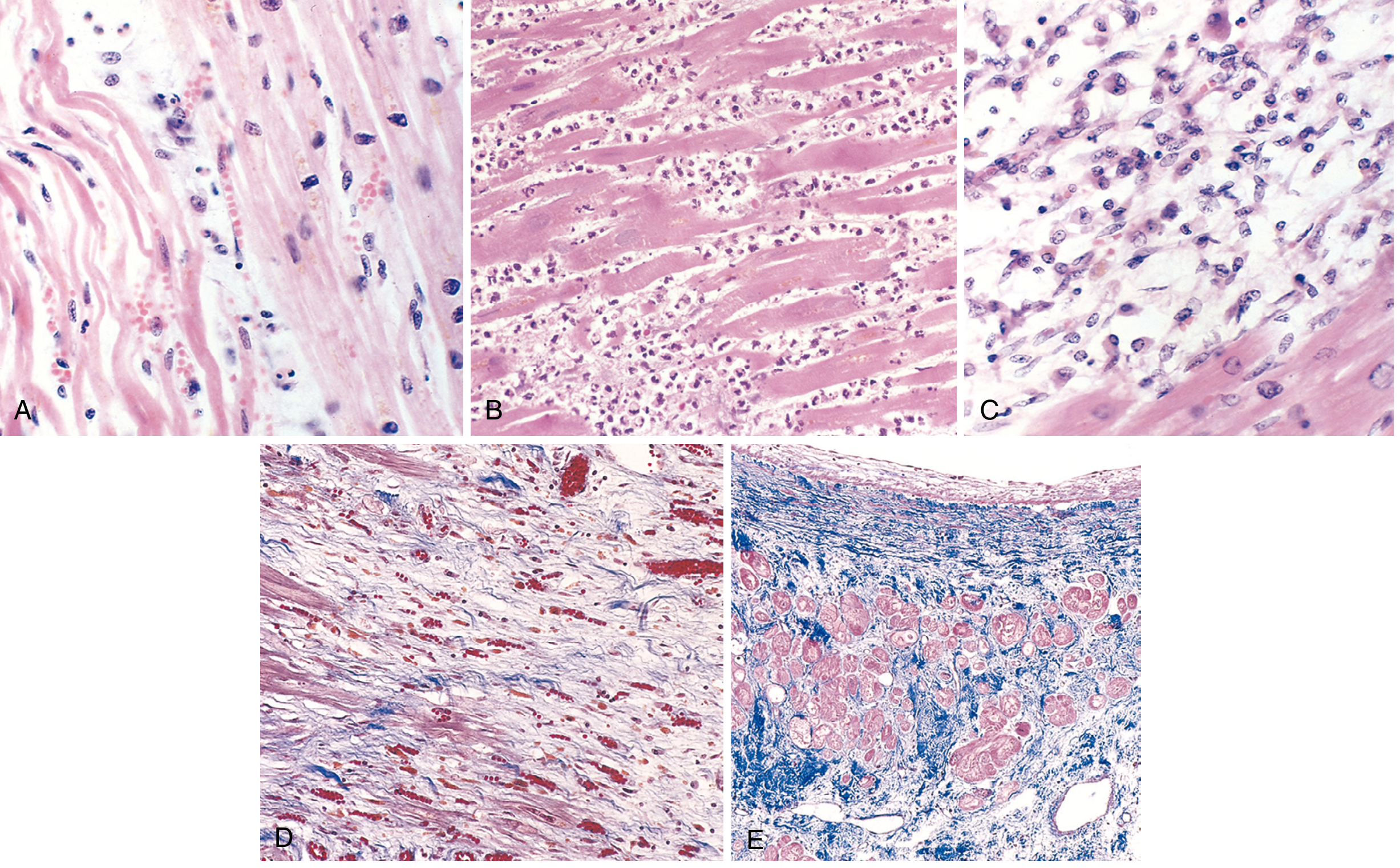
- (A) ~1 day: coagulative necrosis + wavy fibers (elongated, narrow, compared to normal fibers at right), edema fluid, scattered neutrophils
- (B) ~3-4 days: dense neutrophilic infiltrate (peak inflammation)
- (C) ~7-10 days: macrophage phagocytosis clearing necrotic debris
- (D) ~1-2 weeks: granulation tissue - loose collagen + abundant new capillaries (Masson trichrome)
- (E) >6 weeks: Healed scar - dense blue collagen replacing myocardium (Masson trichrome)
Summary: Simplified Clinical Classification
| Stage | Time | Key Feature |
|---|---|---|
| Acute | 0 hrs - 3 days | Coagulative necrosis; neutrophil infiltrate |
| Subacute | 3 days - 6 weeks | Macrophages → granulation tissue → early collagen |
| Old / Healed | >6 weeks (usually >2 months) | Dense collagenous scar; indistinguishable age |
Key Points to Remember
-
Nothing visible grossly or on light microscopy for the first 4-12 hours - the heart looks entirely normal despite lethal ischemia. This is why autopsy diagnosis of very early MI requires TTC staining or electron microscopy.
-
Wavy fiber change - earliest LM finding (30 min to 4 hrs), at infarct borders only.
-
Neutrophils peak at 1-3 days → die off → replaced by macrophages (3-7 days) → granulation tissue (7-10 days).
-
Healing proceeds border → center; large infarcts heal more slowly and incompletely than small ones.
-
Once fully healed (>2 months), the scar cannot be dated - an 8-week scar looks identical to a 10-year scar.
-
Reperfusion (post-thrombolysis/PCI) alters morphology: the infarct becomes hemorrhagic, and irreversibly damaged cells show contraction band necrosis (intense eosinophilic bands of hypercontracted sarcomeres due to calcium influx). Reversibly injured cells are salvaged.
-
In cerebral infarcts, the same principle applies but the timeline differs slightly - liquefactive necrosis dominates (not coagulative), with cavitation eventually forming a cystic space, and the cellular infiltrate includes microglia/macrophages rather than just blood-derived neutrophils.
Source: Robbins & Kumar Basic Pathology, Table 9.2, p. 357 | Robbins, Cotran & Kumar Pathologic Basis of Disease, Fig. 12.12-12.13, p. 514-515
This is a shared conversation. Sign in to Orris to start your own chat.