Heart failure
Reading File
Asking for Preferences
Finding Sources
Searching PubMed
"heart failure" pathophysiology classification
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive content and excellent diagrams. Here is your complete overview:
Heart Failure: Definition, Types, and Pathophysiology
1. Definition
Heart failure (HF) is a clinical syndrome in which the heart cannot meet the metabolic demands of the body at normal filling pressures. It is not a single disease but a final common pathway for many cardiac insults. The preferred term is "heart failure" rather than "congestive heart failure" because pulmonary congestion is not always present - some patients experience dyspnea and reduced exercise capacity without any overt fluid overload.
Normal cardiac output is approximately 5 L/min (for a 70 kg person). In failure, output falls below this - or, in rare cases, is insufficient despite being elevated.
- Textbook of Family Medicine, 9e
- Katzung's Basic & Clinical Pharmacology, 16e
2. Types of Heart Failure
By Ejection Fraction
| Feature | HFrEF | HFpEF |
|---|---|---|
| Ejection fraction | < 45-50% | > 45-50% (normal) |
| Primary defect | Systolic dysfunction - impaired contraction | Diastolic dysfunction - impaired relaxation/filling |
| Mechanism | Ventricle cannot pump effectively; fibers overstretched | Ventricle too stiff/hypertrophied to relax and fill |
| Drug response | Responds well to inotropes | Does NOT respond well to inotropes |
| Prevalence | ~50% of HF cases | ~50% of HF cases |
Both types frequently coexist in the same patient.
- Lippincott Illustrated Reviews: Pharmacology
By Output Level
- Low-output failure (most common): cardiac output is reduced below normal. Typical of MI, cardiomyopathy, hypertension.
- High-output failure (rare): the body's metabolic demands are so excessive that even elevated cardiac output is inadequate. Causes include hyperthyroidism, beriberi (thiamine deficiency), severe anemia, and arteriovenous malformations. This form responds poorly to standard HF drugs and requires treatment of the underlying cause.
By Side Affected
- Left-sided HF: pulmonary congestion, dyspnea, orthopnea
- Right-sided HF: peripheral edema, raised JVP, hepatomegaly
- Biventricular failure: both sides; most chronic HF progresses here
3. Pathophysiology
3a. The Central Problem: LV Remodeling
The modern understanding moves away from a purely hemodynamic model. The central concept is left ventricular (LV) remodeling - progressive dilation and distortion of LV geometry with loss of function. This can be triggered by:
- Coronary artery disease / myocardial infarction
- Hypertension
- Valvular heart disease
- Diabetes, alcoholism, congenital defects, anemia
Remodeling is potentially reversible with appropriate treatment.
At the cellular level, key changes include:
-
Altered calcium flux in myocytes (impaired contraction/relaxation)
-
Down-regulation of beta-adrenergic receptors
-
Shift from fatty acid oxidation to glycolysis
-
Matrix metalloprotease (MMP) activation -> collagen deposition and fibrosis
-
Myocyte apoptosis (programmed cell death)
-
Textbook of Family Medicine, 9e
3b. Neurohormonal Activation (The Vicious Cycle)
Once cardiac output falls, two major compensatory systems activate:
Sympathetic Nervous System (SNS)
- Baroreceptors sense low blood pressure -> vasomotor center increases sympathetic outflow
- Effect: tachycardia, increased contractility, vasoconstriction
- Vasoconstriction increases afterload, which further reduces ejection fraction - creating a downward spiral
- Chronic catecholamine excess is directly toxic to myocytes (via cAMP-dependent calcium overload)
- Beta-adrenergic receptors are progressively down-regulated, reducing sensitivity
Renin-Angiotensin-Aldosterone System (RAAS)
- Low renal blood flow triggers renin release -> angiotensin II -> aldosterone
- Effects: vasoconstriction (more afterload), sodium and water retention (more preload), cardiac fibrosis
- Angiotensin II promotes myocyte apoptosis, hypertrophy, and fibrosis
- Aldosterone "escapes" ACE inhibitor suppression - which is why aldosterone blockers (e.g. spironolactone) are needed in addition to ACE inhibitors/ARBs
Endothelin-1 from dysfunctional endothelium adds further vasoconstriction.
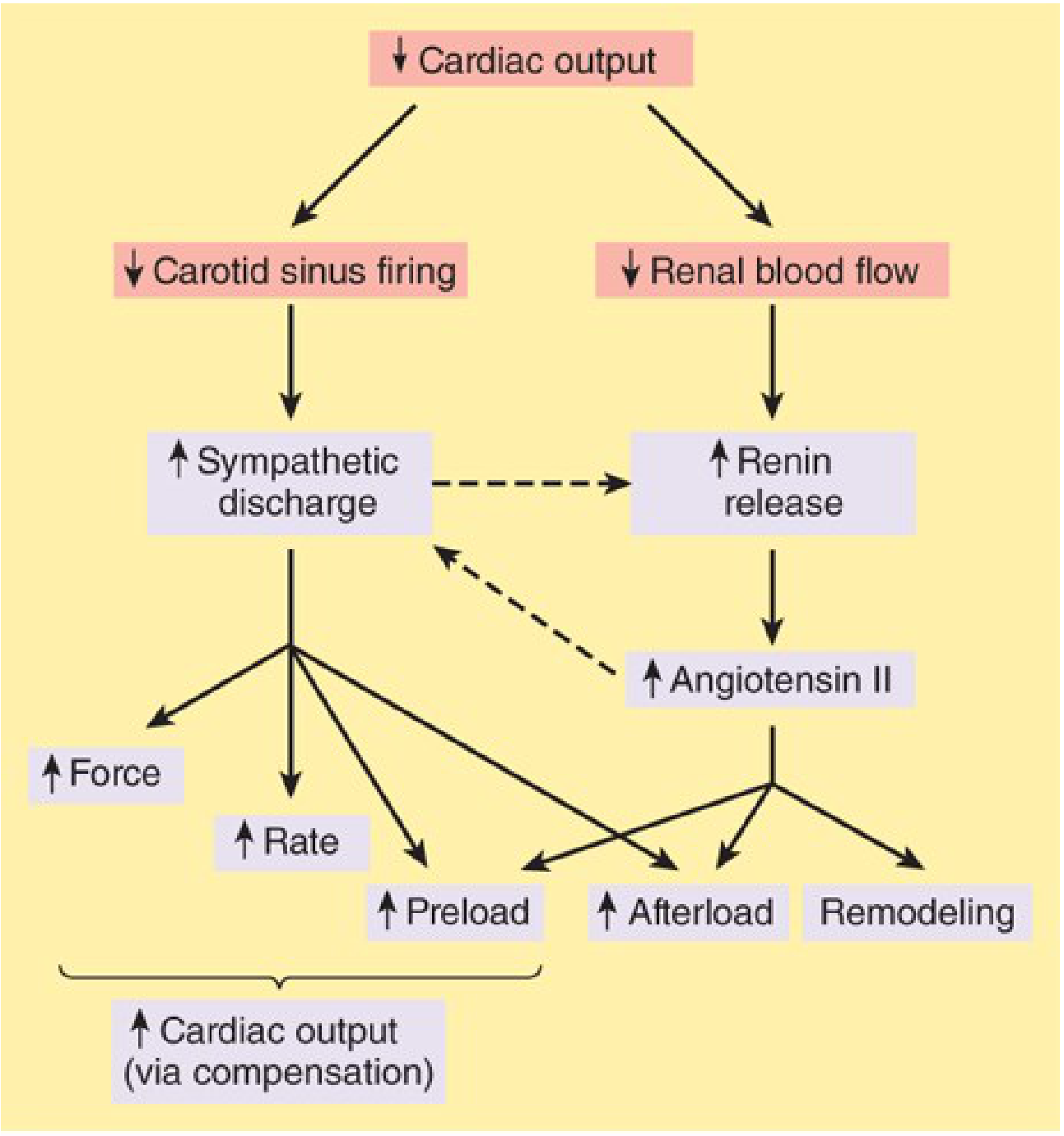
- Katzung's Basic & Clinical Pharmacology, 16e
3c. The Four Major Compensatory Mechanisms
- Increased sympathetic activity: increased heart rate and contractility, vasoconstriction to maintain perfusion pressure. Short-term benefit, long-term harm.
- RAAS activation: sodium/water retention expands blood volume and increases preload (Starling mechanism). If the heart cannot pump the extra volume forward, venous pressure rises and edema results.
- Natriuretic peptides (ANP, BNP): released in response to increased wall stretch. These are the body's counter-regulatory response - they promote vasodilation, natriuresis, and inhibit renin/aldosterone. However, in established HF, the heart becomes resistant to natriuretic peptides (via receptor desensitization and cGMP pathway impairment), blunting this protective effect. This is why BNP levels are actually elevated in HF - the hormone is being produced but not working.
- Myocardial hypertrophy: Initially, wall thickening maintains contractility. Over time, hypertrophy leads to stiffness (diastolic dysfunction) and the fibers become overstretched (systolic dysfunction).
Additional pathological features:
- Inflammation and oxidative stress: tissue hypoperfusion and neurohormonal activation cause mitochondrial dysfunction, cytokine release, and further myocyte death.
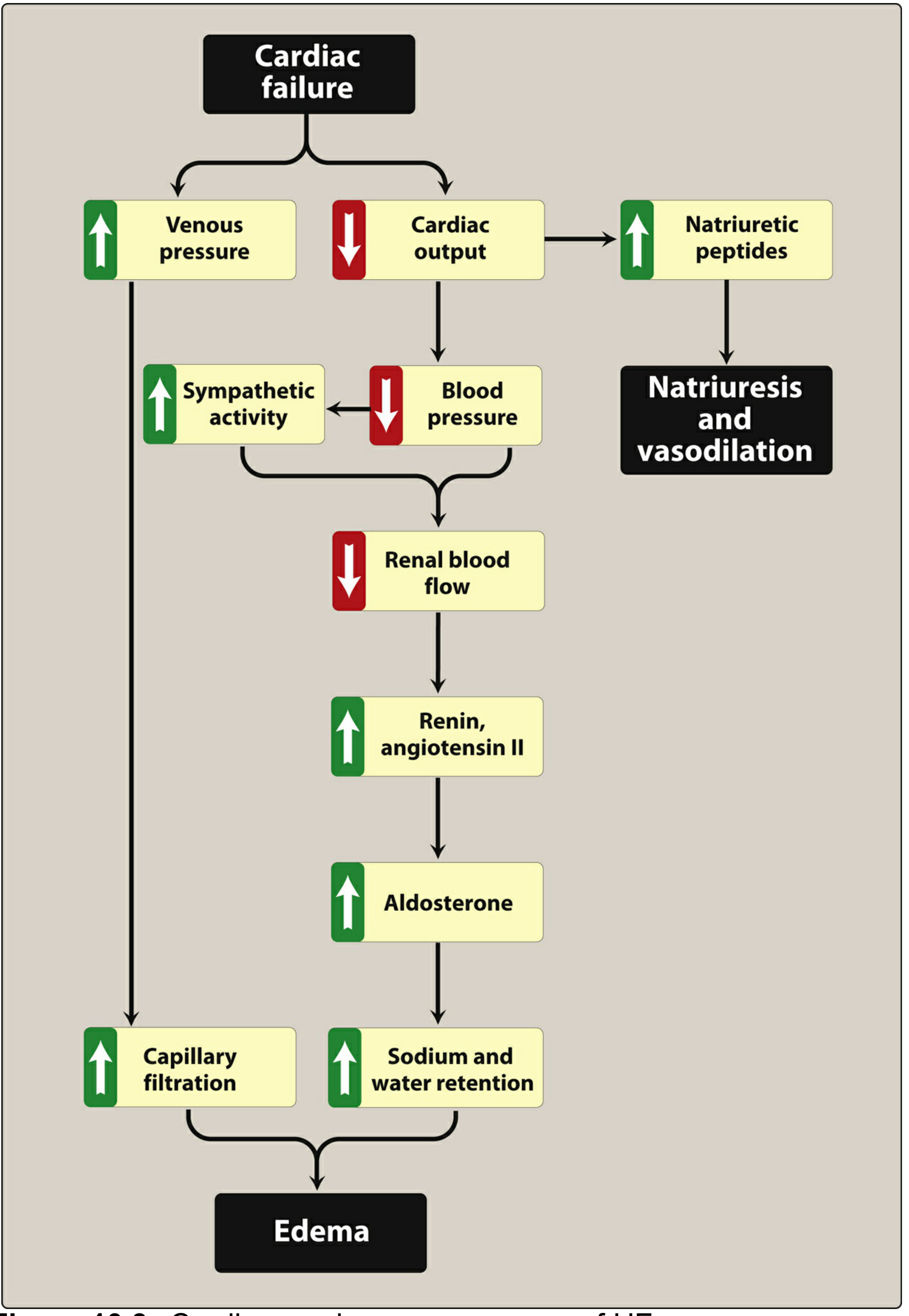
- Lippincott Illustrated Reviews: Pharmacology
4. Classification Systems
NYHA Functional Classification (symptom-based)
| Class | Description |
|---|---|
| I | No symptoms with ordinary activity |
| II | Mild symptoms (dyspnea) with moderate exertion |
| III | Marked limitation; symptoms with minimal activity |
| IV | Symptoms at rest; unable to carry on any activity |
ACC/AHA Staging System (disease progression-based)
This newer system accounts for risk factors and structural disease, not just symptoms. Patients move through stages but cannot move backward (unlike NYHA class, which can improve).
| Stage | Description | Approx. US population |
|---|---|---|
| A | At risk (HTN, DM, IHD, family history) but no LV dysfunction or symptoms | >60 million |
| B | Structural LV dysfunction but asymptomatic (~NYHA I) | ~10 million |
| C | Structural disease + current or prior symptoms (~NYHA II-III) | ~5 million |
| D | Refractory HF, symptoms at rest (~NYHA IV) | ~200,000 |
- Textbook of Family Medicine, 9e
5. Clinical Consequences (Overview)
The primary signs and symptoms stem from two processes:
Forward failure (reduced output):
- Fatigue and decreased exercise tolerance
- Tachycardia
- Cool, clammy extremities
Backward failure (increased filling pressures):
- Left heart: pulmonary congestion, dyspnea, orthopnea, paroxysmal nocturnal dyspnea
- Right heart: peripheral edema, raised JVP, hepatomegaly
Key lab/diagnostic markers: elevated BNP/NT-proBNP (reflect wall stress), ECG changes, cardiomegaly on CXR, reduced EF on echocardiogram.
Summary Diagram of Pathophysiology
Cardiac injury (MI, HTN, valvular disease, etc.)
↓
LV dysfunction + remodeling
↓
↓ Cardiac output
/ \
↑ SNS ↓ Renal blood flow
activation ↓
\ ↑ Renin → ↑ Ang II → ↑ Aldosterone
\ ↓
↑ HR, contractility, vasoconstriction
↑ Na+ and water retention
↓
↑ Preload + ↑ Afterload
↓
Further ↓ EF and ↓ CO
↓
Edema, dyspnea, fatigue
(Symptoms of HF)
Key takeaway for exam purposes: HF pathophysiology is driven by a neurohormonal vicious cycle (SNS + RAAS), not just a pump problem. This is why the most effective treatments - ACE inhibitors, beta-blockers, aldosterone antagonists, and ARNIs - all target this neurohormonal axis rather than simply trying to boost cardiac output.
Sources: Katzung's Basic & Clinical Pharmacology 16e, Lippincott Illustrated Reviews: Pharmacology, Textbook of Family Medicine 9e
This is a shared conversation. Sign in to Orris to start your own chat.