Hey, explain me internuclear ophthalmoplegia, a simple way of explanation so that I can understand the concept properly. You know, whenever I see something related to this MANOphia and all this kind of eye-related questions with the diagram, I'll get panicked too much and I put some wrong answers. So I want some cheat sheet with simple and logical explanation where I can put these answers correct every time I have a step exam. So can you please help me out?
internuclear ophthalmoplegia MLF medial longitudinal fasciculus diagram
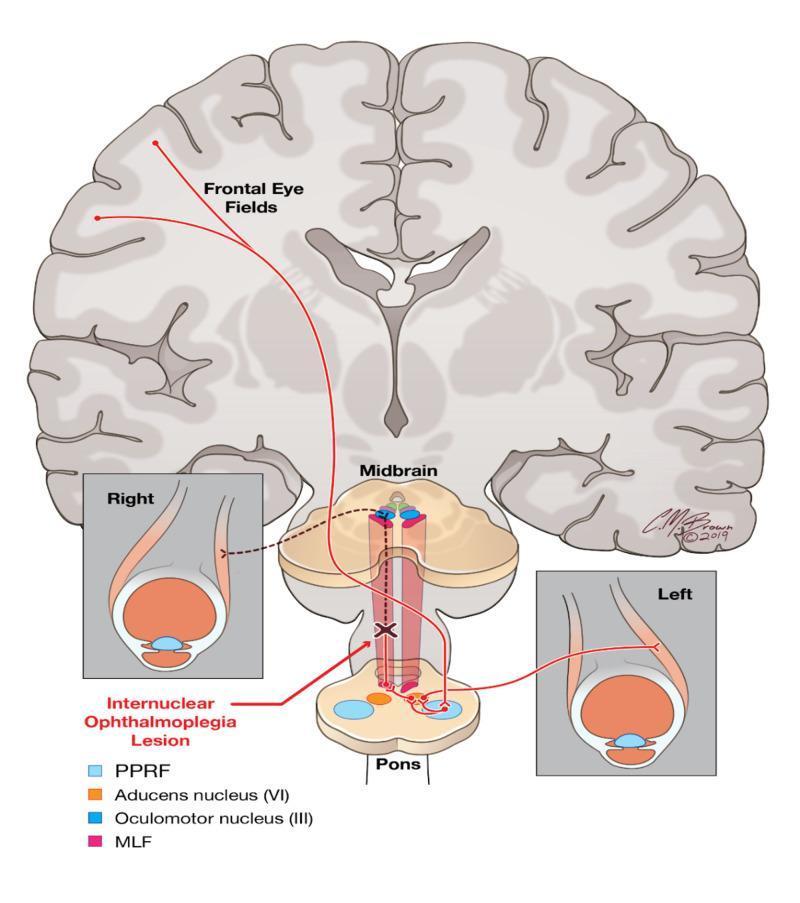
internuclear ophthalmoplegia AND medial longitudinal fasciculus
internuclear ophthalmoplegia
Here’s a simple, Step-style cheat sheet you can memorize and apply fast.
INO in one line
First, decode the pathway (the logic)
- Abducens nucleus (CN VI) activates lateral rectus of one eye
- Through MLF, it sends signal to opposite oculomotor nucleus (CN III) to activate medial rectus of the other eye
What you see on exam
- Right eye should abduct
- Left eye should adduct
- Left eye fails/slows adduction
- Right eye abducts with nystagmus
- This is left INO (named by the weak-adducting side)
Ultra-fast rule to answer MCQs
Classic triad pattern
- Adduction weakness in one eye
- Abducting nystagmus in opposite eye
- Often normal primary gaze (can look straight)
Convergence clue (high-yield)
- Often convergence preserved in isolated INO (helps distinguish from CN III palsy)
- If convergence lost, lesion may be more extensive/rostral
Common causes (Step favorite)
- Young patient: think Multiple sclerosis
- Older patient: think Brainstem ischemic stroke
- Other mimics/causes exist (tumor, trauma, MG mimic), but MS vs stroke is most tested.
How to not confuse with CN III palsy
- No classic ptosis/pupil pattern of CN III palsy
- Problem is internuclear connection (MLF), not the whole third nerve
- Abducting nystagmus in opposite eye is a big clue
Bilateral INO
- Both eyes have adduction deficits on opposite gaze directions
- Can be associated with WEBINO (wall-eyed bilateral INO): bilateral adduction weakness + exotropia pattern
One-and-a-half syndrome (related board trap)
- Lesion hits PPRF/abducens nucleus + MLF on same side
- Result: ipsilateral gaze palsy + ipsilateral INO
- Only movement left: abduction of contralateral eye (often with nystagmus)
20-second exam algorithm
- Is one eye failing to adduct on lateral gaze?
- Is opposite eye abducting with nystagmus?
- If yes -> INO
- Localize lesion to ipsilateral MLF (same side as weak adduction)
- Age clue: young = MS, old = stroke
Memory line
Diagram cue you can visualize
So on the cell membrane of the neurons, we have a receptor called amyloid protein precursor from which it is derived, the receptor. I don't know what this receptor does, but what happens is it is made of a protein. That means it will degrade on a long run and it has to be turnovered. So this, because it is a protein receptor. So this is broken down by alpha secretases and produce some alpha products, which can be easily turned over without any problem and can also occur with beta secretases. It's then broken down into beta products, but beta products are not easy to turn over, hence they accumulate, leading to this A beta or amyloid beta products. This accumulation leads to Alzheimer's disease. Am I right? Make small correction in small intellectual and conceptual reasons.
Hey, explain me the pathophysiology and pathology of Parkinson's disease, where we have the degeneration of gray matter in the deep tissue, especially in the basal ganglia, which will lead to motor problems, I mean movement problems. Technically, we have a communication from cortex to basal ganglia and again from basal ganglia to the cortex, right? So this basal ganglia, when it combines, there is some substantia nigra part, part something, which will release dopamine and this dopamine binds to D1, it stimulates a signal and then it goes to the cortex, but if it binds to D2, it will inhibit the signals something. So moreover, the key factor is that it has to stimulate the cortex with the dopamine. And explain me all the nigrostriatal pathway and Parkinson's disease.
Big picture
- bradykinesia
- rigidity
- resting tremor
- postural instability
Correcting one key conceptual point
It modulates striatal neurons in two pathways:
- D1 receptors (direct pathway): dopamine excites this pathway
- D2 receptors (indirect pathway): dopamine inhibits this pathway
Basal ganglia circuit in simple logic
Normal state (with dopamine)
- Cortex sends excitatory glutamate to striatum.
- SNc dopamine:
- stimulates D1 direct pathway
- inhibits D2 indirect pathway
- Both actions reduce GPi/SNr inhibitory output to thalamus.
- Thalamus can excite motor cortex.
- Movement is facilitated.
Parkinson state (dopamine loss)
- Direct pathway underactive (less D1 drive).
- Indirect pathway overactive (loss of D2 inhibition).
- GPi/SNr become overactive.
- Thalamus is excessively inhibited.
- Less thalamocortical excitation -> hypokinesia/bradykinesia.
Nigrostriatal pathway (what to remember)
- Origin: Substantia nigra pars compacta
- Projection: Striatum (caudate + putamen)
- Neurotransmitter: Dopamine
- Function: fine-tunes motor output via direct and indirect pathways
- Degeneration here is central to PD motor features
Pathology of Parkinson disease
- Depigmentation (pale substantia nigra, locus coeruleus)
- Neuronal loss and gliosis in SNc
- Lewy bodies (intracytoplasmic eosinophilic inclusions)
- Major protein in Lewy bodies: alpha-synuclein (misfolded/aggregated), with ubiquitin
- often begins in lower brainstem/olfactory structures, then midbrain/cortex
- helps explain early non-motor symptoms
Why non-motor symptoms happen
- hyposmia
- constipation
- REM sleep behavior disorder
- depression/anxiety
- autonomic dysfunction
- cognitive decline later (in some patients)
One-line exam summary
And where is the substation like the power compact present? I forgot, I mean, I've studied long back. I couldn't remember. Can you explain to me like a first year med student?
- It is in the midbrain (mesencephalon), part of the brainstem.
- It lies in the ventral midbrain, just above the cerebral peduncles.
- It is one part of substantia nigra:
- Pars compacta (SNc) = dopamine neurons (the one affected in Parkinson disease)
- Pars reticulata (SNr) = output nucleus, more GABA-type function
First-year mental map
and dopamine support comes from a “battery pack” in midbrain:
This is the nigrostriatal pathway.
Quick memory trick
yes plase
1) Where SNc is (location sketch)
Side view (simplified)
[Cerebrum]
|
[Thalamus]
|
[Midbrain] <-- Substantia nigra is here
├─ SNc (pars compacta) = dopamine neurons
└─ SNr (pars reticulata)
|
[Pons]
|
[Medulla]
2) Nigrostriatal pathway (key arrow)
SNc (midbrain) --dopamine--> Striatum (caudate + putamen)
3) Motor loop in one glance
Cortex -> Striatum -> GPi/SNr -> Thalamus -> Cortex
^
|
Dopamine from SNc
- activating D1 (direct pathway)
- inhibiting D2 (indirect pathway)
4) Parkinson version (one-liner)
SNc neurons degenerate -> less dopamine in striatum -> movement becomes hard
5) 10-second memory hook
Battery low -> slow movement (Parkinsonian bradykinesia).
now all together compact usmle topper ist ranker notes short and hy compact
Parkinson Disease + Nigrostriatal Pathway (Ultra-Compact USMLE Notes)
1) Core lesion
- Degeneration of dopaminergic neurons in Substantia Nigra pars compacta (SNc)
- Location: ventral midbrain (brainstem), above cerebral peduncles
- Result: ↓ dopamine in striatum (putamen > caudate)
2) Basal ganglia motor loop
- D1 (direct pathway): stimulated by dopamine -> pro-movement
- D2 (indirect pathway): inhibited by dopamine -> pro-movement
3) Parkinson pathophysiology
- ↓ D1 activation -> weaker direct pathway
- ↓ D2 inhibition -> stronger indirect pathway
- -> ↑ GPi/SNr inhibition of thalamus
- -> ↓ thalamocortical motor drive
- -> bradykinesia/hypokinesia
4) Classic clinical features
- Bradykinesia (required for diagnosis with other features)
- Resting tremor (“pill-rolling”)
- Cogwheel/lead-pipe rigidity
- Postural instability, shuffling gait, masked facies, micrographia
5) Pathology
- Pale substantia nigra (depigmentation)
- Lewy bodies (intracytoplasmic eosinophilic inclusions)
- Main protein: alpha-synuclein (misfolded), ubiquitin-positive
6) High-yield non-motor symptoms (often early)
- Hyposmia
- Constipation
- REM sleep behavior disorder
- Depression/anxiety
- Autonomic dysfunction
- Cognitive decline later
7) Must-know compare
- Parkinson disease: basal ganglia disorder, no UMN weakness pattern
- Nigrostriatal tract: SNc -> striatum dopamine pathway (key damaged pathway)
8) 1-line exam formula
9) Memory hook
what is gpi?nsr
- GPi = Globus Pallidus internus (internal segment of globus pallidus)
- SNr = Substantia Nigra pars reticulata
They send mostly GABA (inhibitory) signals to the thalamus (and brainstem targets).
- More GPi/SNr firing -> more thalamus inhibition -> less movement
- In Parkinson disease, GPi/SNr output becomes relatively overactive, so movement decreases.
How this soleus muscle is different from all other muscles, which will is very much used to blood glucose levels fast, so the soleus push-up can be very much helpful in diabetes patients.
Why soleus is different
- Mostly type I (slow-twitch, oxidative) fibers
- Built for endurance/posture (active all day in standing/walking)
- High mitochondrial and capillary density
- Can do long, low-intensity contractions with low fatigue
About “soleus push-up” and glucose
- Small studies suggest seated soleus contractions may improve post-meal glucose handling acutely.
- Mechanism: muscle contraction stimulates glucose uptake (partly insulin-independent pathways like GLUT4 translocation).
- It is a useful adjunct movement snack, especially for sedentary periods.
But important correction
- It is not superior to all exercise for diabetes control.
- It does not replace walking, resistance training, weight loss strategy, meds, sleep, and diet quality.
- Evidence is promising but still limited compared with standard diabetes exercise recommendations.
Practical use (safe approach)
- During long sitting: 3-5 min every 30-60 min
- After meals: gentle calf raises/soleus pulses for 10-20 min (as tolerated)
- Keep doing full-body aerobic + resistance exercise weekly
Give me a small short note on trinucleotide repeats and augmentation of the disease in the progeny. Like if the grandfather gets like at 60 years, the father will get like around maybe 55 years, the son will get maybe 50 years, you know, something like that. There will be augmentation happening. That line. Am I right? Correct me and make a short note.
The correct term is anticipation (not augmentation).
Short note: Trinucleotide repeat disorders and anticipation
- Some diseases are caused by expansion of repeated 3-base DNA sequences (eg, CAG, CTG, CGG).
- During meiosis, repeat length can increase in the next generation.
- More repeats -> earlier onset and often more severe disease.
- This phenomenon is called anticipation.
- Grandfather onset at 60
- Father at 55
- Son at 50
This pattern suggests anticipation due to repeat expansion.
Classic examples
- Huntington disease (CAG, paternal expansion tendency)
- Myotonic dystrophy type 1 (CTG, often maternal expansion)
- Fragile X syndrome (CGG expansion in FMR1; premutation -> full mutation across generations)
Hey ChatGPT, I've heard that corneal transplant is the number one transplantation with least rejection rate because there is no, it is like, it's like a privileged organ where there will be decreased immune surveillance. Hence, it's the most acceptable transplantation, right? But I've studied somewhere that CJD or Creutzfeldt-Jakob disease can all occur very rarely during corneal transplant because of this beta 2 prion exposure where it will get converts in our normal alpha configuration to beta and it will lead to accumulation and damage of neuronal myelins leading to this intracellular vacuoles. But if there is, how come it's like the retinal barrier or blood-brain barrier or what do you say, the eye-brain retinal barrier is very least, has the least immune surveillance and something like that. Explain to me.
1) Why corneal graft rejection is low
- No blood/lymph vessels in normal cornea
- Low MHC expression in corneal tissues
- Local immunoregulatory factors in aqueous humor and anterior chamber
- ACAID-type immune tolerance mechanisms
2) Why infection transmission (eg, prion) can still occur
- Rejection = recipient immune attack on donor graft
- Transmission = donor tissue carries infectious agent into recipient
3) Corneal transplant and CJD
- Very rare iatrogenic transmission has been reported historically with corneal grafts.
- It is not because of “beta-2 prion.” The key is misfolded prion protein (PrPSc) inducing misfolding of normal PrPC.
- Prion diseases cause spongiform change (vacuolation), neuronal loss, gliosis; they are not primarily a “myelin damage” disorder.
- Modern donor screening has made this risk extremely low.
4) Barrier confusion (eye vs brain)
- Blood-retinal barrier and blood-aqueous barrier regulate trafficking into ocular compartments.
- Blood-brain barrier is CNS-specific.
- Corneal immune privilege is mostly local ocular immunobiology, not simply “same as BBB.”
One-line summary
ptt normal
- PT/INR = extrinsic pathway
- aPTT = intrinsic + common pathway
dizziness on anticogulations
- severe or sudden headache
- new weakness, numbness, trouble speaking, vision change
- fainting, chest pain, shortness of breath
- black stools, vomiting blood, blood in urine
- recent fall/head injury
- persistent worsening dizziness
- occult bleeding -> anemia/hypotension
- over-anticoagulation (if warfarin, high INR)
- dehydration, vestibular issues, arrhythmia, etc.
- do not take extra doses
- check your medication name/dose and timing errors
- if on warfarin, get INR checked promptly
- contact your prescriber today for same-day advice
rivoraxaban is what
It is a direct factor Xa inhibitor.
- prevent stroke in nonvalvular atrial fibrillation
- treat/prevent DVT and PE
- clot prevention after some surgeries
- fixed dosing (usually no routine INR monitoring like warfarin)
- main risk: bleeding
- take exactly as prescribed (some doses must be taken with food)
A 17-year-old right-handed boy is brought to the emergency department due to brief loss of consciousness after colliding with another player during a football game. The patient reports "fogginess" but has had no headache, vomiting, focal weakness or numbness, or seizures. He has no other medical conditions and takes no medications. Vital signs are within normal limits. On physical examination, the patient demonstrates retrograde amnesia about the game but no focal neurologic deficits. CT angiography of the head reveals no skull fractures or intracranial bleeding, but a small lesion in the left temporoparietal region composed of abnormal vascular channels is noted and is shown in the exhibit. Without treatment of this abnormality, which of the following will most likely occur in this patient? A. Brain abscess (2%) B. Flaccid paralysis (2%) C. Malignant transformation (2%) D. Spontaneous hemorrhage (83%) E. Spontaneous resolution (8%) his young patient most likely sustained a concussion. Because he lost consciousness and has retrograde amnesia, imaging is performed to evaluate for intracranial hemorrhage. Incidentally, the CT angiogram reveals a cluster of dilated vessels (ie, "bag of worms") in the left temporoparietal region with large arterial feeder vessels. This is consistent with an arteriovenous malformation (AVM). AVMs occur when an artery directly anastomoses with veins without an interposed capillary bed. The resulting high-pressure blood flow through weak vessels (ie, veins) predisposes patients to aneurysm formation and/or spontaneous hemorrhage. Seizures also commonly develop and may be due to local accumulation of hemosiderin, a cortical irritant, from microbleeds. Most AVMs are congenital and sporadic. They can occasionally occur as a component of hereditary hemorrhagic telangiectasia (ie, Osler-Weber-Rendu syndrome), an autosomal dominant disorder associated with mucocutaneous telangiectasias (eg, nasal mucosa, intestinal mucosa) and multiorgan AVMs (eg, lungs, liver, brain). (Choice A) The pulmonary capillaries filter potential debris from the venous blood (eg, clots, bacteria), preventing it from entering the systemic circulation. A pulmonary AVM (but not a cerebral AVM) bypasses this filtering system, leading to paradoxic embolization into the systemic arteries (eg, stroke or brain abscess). (Choice B) Flaccid paralysis can occur with lesions that affect the peripheral nervous system. However, lesions in the CNS lead to upper motor neuron signs (eg, spastic paralysis, hyperreflexia). In addition, cerebral AVMs very rarely lead to motor deficits. (Choice C) AVMs are congenital malformations rather than neoplastic proliferations. Therefore, they are not at risk of malignant transformation. (Choice E) Although some cerebral AVMs are stable and do not lead to either seizures or intracerebral hemorrhage, they never resolve spontaneously. Educational objective: Arteriovenous malformations occur when an artery directly anastomoses with veins without an interposed capillary bed. They may result in spontaneous intracerebral hemorrhage or seizures, or they may be discovered incidentally when brain imaging is obtained for another reason Convert this UWorld question into ultra high-yield USMLE notes: 1. Diagnosis: 2. Key clue from question stem (1 line): 3. Why correct answer (max 2 lines): 4. Why others wrong (1 line each): 5. Buzzword trigger: 6. Trap / trick tested: 7. One-liner memory hook: Make it extremely concise. No paragraphs.
-
Diagnosis:
- Cerebral arteriovenous malformation (AVM) (incidental finding after concussion workup)
-
Key clue from question stem (1 line):
- Young patient + CTA showing “bag of worms” abnormal vascular channels with arterial feeders in brain.
-
Why correct answer (max 2 lines):
- AVM = direct artery-to-vein shunt without capillary bed.
- High-flow stress in fragile veins -> highest risk: spontaneous intracranial hemorrhage (also seizures).
-
Why others wrong (1 line each):
- A Brain abscess: classic with pulmonary AVM (bypasses lung filter), not cerebral AVM.
- B Flaccid paralysis: LMN/peripheral lesion finding; cerebral lesions cause UMN pattern.
- C Malignant transformation: AVM is congenital vascular malformation, not neoplasm.
- E Spontaneous resolution: AVMs generally persist; do not reliably regress spontaneously.
-
Buzzword trigger:
- “Bag of worms” intracranial vessels on angiography in a young person.
-
Trap / trick tested:
- Incidental AVM found during trauma evaluation: question asks natural history if untreated, not concussion management.
- Confusion between cerebral AVM vs pulmonary AVM complications.
-
One-liner memory hook:
- “No capillary buffer -> arterial pressure hits veins -> bleed risk.”
A 4-year-old boy is evaluated for difficulty walking. Past medical history includes frequent respiratory infections. Cultured cells from this patient demonstrate a high rate of radiation-induced genetic mutation. This patient is most likely to experience which of the following? taxia-telangiectasia is an autosomal recessive disorder. Cerebellar atrophy leads to the ataxia that occurs in the first years of life. (Oculocutaneous telangiectasia is another manifestation, but is usually delayed. Telangiectasias are abnormal dilatations of capillary vessels.) Patients with ataxia-telangiectasia also have severe immunodeficiency with repeated sinopulmonary infections. The risk of cancer in these patients is increased significantly because of inefficient DNA repair. DNA damage and repair is an ongoing process. A lot of naturally-occurring agents cause DNA damage: sunlight, UV light, ionizing radiation (gamma and X-ray), and free oxygen radicals are produced during the cell cycle. Some chemicals (hydrocarbons in cigarette smoke, aflatoxins produced by moldy peanuts) are also harmful to DNA. DNA repair occurs continuously by means of direct reversal of damage, base and nucleotide excision repair, and mismatch repair. A number of inherited disorders are caused by deficient DNA-repair enzymes: 1. Ataxia-telangiectasia is characterized by DNA hypersensitivity to ionizing radiation. 2. In xeroderma pigmentosum, DNA is hypersensitive to UV radiation, causing premature skin aging and increased risk of skin cancer (malignant melanoma and squamous cell carcinoma). 3. Fanconi anemia is caused by hypersensitivity of DNA to cross-linking agents. 4. Bloom syndrome is characterized by generalized chromosomal instability. Increased susceptibility to neoplasms is present. 5. Hereditary nonpolyposis colorectal cancer (HNPCC) occurs due to a defect in DNA mismatch-repair enzymes. It leads to increased susceptibility to colon cancer. (Choice A) Neurofibrillary tangles in the neocortex are seen in Alzheimer disease. (Choice C) Loss of neurons in the substantia nigra is characteristic of Parkinson disease. (Choice D) Atrophy of caudate nucleus is seen in Huntington disease. (Choice E) Posterior column involvement is seen with both vitamin B 12 deficiency and syphilis. (Choice F) The presence of Lewy bodies is characteristic of Parkinsonism. Educational Objective: Ataxia-telangiectasia is an autosomal-recessive disorder resulting from a defect in DNA-repair genes. The DNA of these patients is hypersensitive to ionizing radiation. Manifestations include cerebellar ataxia, oculocutaneous telangiectasias, repeated sinopulmonary infections, and an increased incidence of malignancy.
-
Diagnosis:
- Ataxia-telangiectasia (ATM mutation, AR)
-
Key clue from stem (1 line):
- Child with early ataxia + recurrent sinopulmonary infections + radiation-induced DNA damage.
-
Why correct answer (max 2 lines):
- ATM defect -> impaired double-strand DNA break repair -> hypersensitivity to ionizing radiation.
- Causes cerebellar degeneration and markedly increased risk of lymphoid malignancy.
-
Why others wrong (1 line each):
- Alzheimer (neurofibrillary tangles): degenerative dementia in elderly, not pediatric DNA-repair syndrome.
- Parkinson (SN degeneration/Lewy bodies): adult movement disorder, unrelated to ATM immunodeficiency pattern.
- Huntington (caudate atrophy): AD trinucleotide disease with chorea, not recurrent infections/radiosensitivity.
- Posterior column disease (B12/tabes): sensory ataxia mechanism, not AR DNA-repair defect in child.
- Lewy body parkinsonism: alpha-synucleinopathy in older adults, not childhood cerebellar ataxia syndrome.
-
Buzzword trigger:
- “Ataxia + telangiectasia + recurrent infections + radiosensitivity”
-
Trap / trick tested:
- Differentiate cerebellar ataxia from neurodegenerative adult disorders; link recurrent infections to DNA-repair/immunodeficiency.
-
One-liner memory hook:
- “A-T = ATM broken -> can’t repair radiation DNA breaks -> Ataxia, Telangiectasia, Infections, Tumor risk.”