Lipid lowering drugs(statin) and Anticoagulants. Give me detailed pharmacological notes on these 2 topics
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive content from multiple authoritative textbooks. Let me compile the full pharmacological notes.
Pharmacological Notes: Lipid-Lowering Drugs (Statins) & Anticoagulants
PART 1: HMG-CoA REDUCTASE INHIBITORS (STATINS)
1. Classification & Available Agents
Statins are the cornerstone of pharmacological lipid-lowering therapy. They are classified by their LDL-C lowering potency:
| Intensity | Drugs & Daily Doses | Expected LDL-C Reduction |
|---|---|---|
| High-intensity | Atorvastatin 40–80 mg; Rosuvastatin 20–40 mg | ≥50% |
| Moderate-intensity | Atorvastatin 10–20 mg; Rosuvastatin 5–10 mg; Simvastatin 20–40 mg; Pravastatin 40–80 mg; Lovastatin 40–80 mg; Fluvastatin XL 80 mg; Pitavastatin 1–4 mg | 30–<50% |
| Low-intensity | Fluvastatin 20–40 mg; Lovastatin 20 mg; Pravastatin 10–20 mg; Simvastatin 10 mg | <30% |
(Source: 2018 AHA/ACC Guidelines; Goodman & Gilman, Table 37-7)
2. Mechanism of Action
Primary mechanism: Statins competitively and reversibly inhibit HMG-CoA reductase (3-hydroxy-3-methylglutaryl coenzyme A reductase), the rate-limiting enzyme in cholesterol biosynthesis. This enzyme catalyzes the conversion of HMG-CoA → mevalonate, an early committed step in the mevalonate pathway.
Downstream consequences:
- ↓ intrahepatic cholesterol → upregulation of hepatic LDL receptors (via SREBP activation) → increased clearance of LDL-C, IDL, and VLDL remnants from plasma
- ↓ VLDL synthesis → secondary reduction in triglycerides
- Modest ↑ HDL-C (mechanism not fully elucidated)
Pleiotropic effects (beyond lipid lowering):
- Anti-inflammatory (↓ CRP, cytokines)
- Plaque stabilization
- Improved endothelial function (↑ nitric oxide bioavailability)
- Antithrombotic properties
3. Pharmacokinetics
| Property | Detail |
|---|---|
| Oral absorption | Variable: 30%–85% |
| Prodrugs | Simvastatin and lovastatin are inactive lactones; activated in the liver to β-hydroxy acid forms |
| Active forms | All other statins administered as active β-hydroxy acids |
| First-pass hepatic uptake | Extensive — mediated by OATP1B1 (organic anion transporter); ensures hepatic selectivity |
| Systemic bioavailability | 5%–30% (high hepatic extraction) |
| Half-life | Most: ~4 h; Atorvastatin and Rosuvastatin: longer (14–19 h) |
| Dosing time | Short t½ statins → take in the evening (hepatic cholesterol synthesis peaks midnight–2 AM); Atorvastatin/Rosuvastatin → any time |
CYP Metabolism:
- CYP3A4/3A5: Atorvastatin, Lovastatin, Simvastatin
- CYP2C9: Fluvastatin (50–80%), Rosuvastatin (~10%)
- Not CYP-metabolized: Pravastatin (excreted unchanged) — lowest drug interaction risk
(Goodman & Gilman's Pharmacological Basis of Therapeutics)
4. Therapeutic Uses / Indications
- Primary prevention: Patients with elevated 10-year ASCVD risk ≥7.5% (age 40–75)
- Secondary prevention: Established ASCVD (MI, stroke, peripheral arterial disease)
- Familial hypercholesterolemia (heterozygous and homozygous forms)
- Diabetes mellitus (high cardiovascular risk)
- Post-ACS: High-intensity statin regardless of baseline LDL
- Stroke prevention (secondary)
- Effective in both men and women
The lipid-lowering effect appears within the first week and stabilizes after ~4 weeks.
5. Adverse Effects
| Adverse Effect | Notes |
|---|---|
| Myalgia/muscle pain | Most common cause of discontinuation (5–10%); dose-dependent; may occur without ↑CK; risk ↑ with age, renal impairment, hypothyroidism |
| Myopathy/rhabdomyolysis | Rare but serious; ↑ risk with drug interactions (CYP3A4 inhibitors, fibrates) |
| Hepatotoxicity | 1–2% asymptomatic aminotransferase elevations (>3×ULN); routine liver monitoring not required; statin hepatotoxicity is not increased in chronic hepatitis C or hepatic steatosis |
| New-onset diabetes | Small but real increased risk; net cardiovascular benefit usually outweighs |
| GI symptoms | Abdominal pain, diarrhea, bloating, constipation |
| CNS | No evidence of negative impact on cognitive function |
Management of myalgias:
- Discontinue statin; evaluate for hypothyroidism, renal/hepatic disease, vitamin D deficiency
- Myalgias typically resolve within 2 months of stopping
- Retry same or lower dose, or switch to a different statin
(Washington Manual of Medical Therapeutics)
6. Drug Interactions
Pharmacokinetic interactions (CYP3A4 inhibitors — ↑ statin levels → ↑ myopathy risk):
- Azole antifungals (itraconazole, ketoconazole)
- Macrolide antibiotics (erythromycin, clarithromycin)
- Cyclosporine
- HIV protease inhibitors
- Amiodarone, diltiazem, verapamil, amlodipine
- Nefazodone
- Grapefruit juice (large quantities)
Pharmacodynamic interactions:
- Gemfibrozil (greatest risk): inhibits OATP1B1 uptake AND CYP2C8, nearly doubling statin plasma concentrations → high rhabdomyolysis risk
- Niacin: additive skeletal muscle cholesterol synthesis inhibition
Specific simvastatin limits:
- Max 20 mg with amlodipine or amiodarone
- Max 10 mg with diltiazem or verapamil
- Contraindicated with cyclosporine, HIV protease inhibitors, erythromycin, gemfibrozil
Other interactions:
- Simvastatin and Rosuvastatin may ↑ warfarin levels (monitor INR)
7. Contraindications
- Pregnancy and lactation (absolute contraindication)
- Active liver disease or persistent unexplained elevation of transaminases
- Hypersensitivity to any component
- Concurrent gemfibrozil use with most statins
8. Pharmacological Note: Hepatic Transporter (OATP1B1)
The efficient first-pass hepatic uptake by OATP1B1 is critical to statin pharmacology — it concentrates statins in the liver (target organ) while minimizing systemic levels, thereby reducing muscle adverse effects. Genetic polymorphisms of OATP1B1 (e.g., SLCO1B1 variants) can impair transporter function, reducing hepatic uptake and increasing systemic statin exposure → ↑ myopathy risk.
PART 2: ANTICOAGULANTS
Overview
Anticoagulants prevent formation and extension of thrombi. They are classified into parenteral and oral agents.
A. PARENTERAL ANTICOAGULANTS
1. Unfractionated Heparin (UFH)
Source: Heterogeneous mixture of negatively charged polysulfated glycosaminoglycans from porcine intestine or bovine lung; MW range 3,000–30,000 Da.
Mechanism of Action:
- Binds antithrombin (AT) via a specific pentasaccharide sequence → induces conformational change → dramatically enhances AT's ability to inhibit:
- Thrombin (factor IIa) — requires pentasaccharide + chain length ≥18 sugar units
- Factor Xa — requires pentasaccharide sequence only
- Other serine proteinases (IXa, XIa, XIIa)
- ⚠️ Up to two-thirds of heparin chains lack the pentasaccharide sequence and have minimal anticoagulant activity
Dosing:
- Treatment: 80 U/kg IV bolus → 18 U/kg/hr infusion (titrate to aPTT)
- Prophylaxis: 5,000 U SC 2–3 times daily
Monitoring: aPTT (target 60–100 sec); anti-factor Xa activity; ACT for cardiac procedures
Reversal: Protamine sulfate (1 mg per 100 U UFH, IV slowly)
Adverse Effects:
- Bleeding (major risk)
- Heparin-induced thrombocytopenia (HIT): Immune-mediated (IgG antibodies to PF4-heparin complex) → paradoxical thrombosis; thrombocytopenia typically day 5–14
- Osteoporosis (long-term use)
- Hypoaldosteronism
2. Low-Molecular-Weight Heparins (LMWHs)
Examples: Enoxaparin, Dalteparin, Nadroparin, Tinzaparin
Produced by: Chemical or enzymatic depolymerization of UFH; chain length 4–22 monosaccharide units
Mechanism:
- Primarily enhances AT-mediated inhibition of factor Xa
- Less effect on thrombin (shorter chains cannot bridge AT to thrombin)
- Higher anti-Xa : anti-IIa ratio vs. UFH
Advantages over UFH:
- Predictable dose-response (reduced plasma protein and endothelial binding)
- Higher bioavailability after SC injection
- Longer half-life (3–5 h)
- Renal excretion
- Does not cross the placenta → safe in pregnancy
- Significantly less risk of HIT than UFH
Dosing (Enoxaparin as example):
- Treatment VTE: 1 mg/kg SC q12h (or 1.5 mg/kg once daily)
- Prophylaxis: 40 mg SC daily (general surgery); 40 mg SC daily or 30 mg SC bid (orthopedic)
- ACS: 1 mg/kg SC q12h
| Agent | Plasma t½ (min) |
|---|---|
| Enoxaparin | 130–180 |
| Dalteparin | 120–139 |
| Nadroparin | 130–162 |
Monitoring: Usually not required; anti-Xa levels in renal impairment, obesity, pregnancy
Reversal: Protamine sulfate (partial reversal only)
Special note: Do not use LMWH acutely in patients who develop HIT on UFH (cross-reactive antibodies risk)
3. Fondaparinux
Nature: Synthetic pentasaccharide (the exact AT-binding sequence of heparin)
Mechanism:
- Binds antithrombin with high affinity → catalytic inhibition of factor Xa only
- No effect on thrombin (chain too short to bridge AT to thrombin)
Pharmacokinetics:
- SC injection once daily
- Half-life: ~17 h
- Renal excretion (avoid if CrCl <30 mL/min)
Dosing:
- Treatment: 7.5 mg SC daily (5 mg if <50 kg; 10 mg if >100 kg)
- Prophylaxis: 2.5 mg SC daily (avoid if <50 kg)
Advantages: Does not cause HIT (does not bind PF4)
Monitoring: Anti-Xa activity (if needed)
Reversal: No specific antidote; recombinant factor VIIa may be used
4. Direct Thrombin Inhibitors (DTIs) — Parenteral
| Property | Bivalirudin | Argatroban |
|---|---|---|
| MW | 1,980 Da | 527 Da |
| Thrombin binding | Active site + exosite 1 | Active site only |
| t½ | ~25 min | ~45 min |
| Elimination | Enzymatic (blood) + renal | Hepatic metabolism |
| Monitoring | aPTT, dilute thrombin time | aPTT |
| Key indication | PCI (alternative to heparin) | HIT |
| Renal clearance | Partial | No |
Mechanism: Direct, AT-independent inhibition of thrombin
Argatroban: Drug of choice for HIT requiring anticoagulation; dose: 2 µg/kg/min IV (start); hepatically metabolized → used in renal failure
Bivalirudin: Used for PCI; also in HIT patients requiring cardiac procedures
B. ORAL ANTICOAGULANTS
5. Warfarin (Vitamin K Antagonist — VKA)
Mechanism of Action:
All vitamin K-dependent clotting factors (II, VII, IX, X, and anticoagulant proteins C and S) require γ-carboxylation of glutamic acid residues at their N-termini. This modification enables calcium-dependent binding to phospholipid surfaces (essential for coagulation activity).
The process:
- Dietary vitamin K → reduced to vitamin K hydroquinone (by vitamin K reductase)
- Vitamin K hydroquinone acts as cofactor for γ-glutamyl carboxylase → γ-carboxylation of clotting factor precursors
- During this process, vitamin K hydroquinone is oxidized → vitamin K epoxide
- Vitamin K epoxide is recycled back to vitamin K by vitamin K epoxide reductase (VKOR)
Warfarin blocks VKOR → depletes reduced vitamin K → clotting factors synthesized are only partially γ-carboxylated → biologically inactive
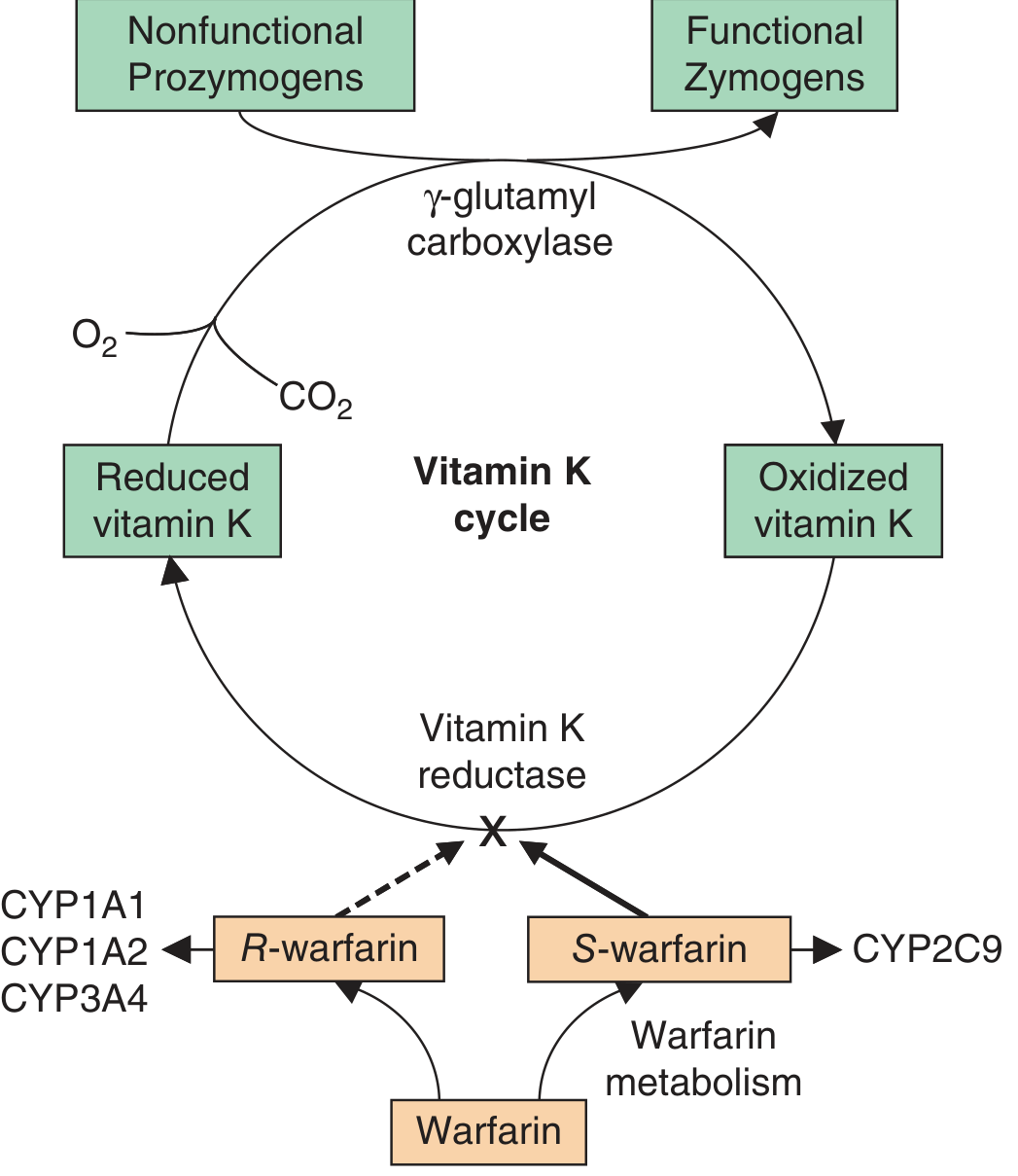
Factors affected: II (prothrombin), VII, IX, X; Proteins C and S
Pharmacokinetics:
- Racemic mixture of S and R enantiomers; S-warfarin is more potent (5× more active)
- Rapidly and almost completely absorbed orally; peak blood levels ~90 min
- Plasma t½: 36–42 h
-
97% protein-bound (albumin); only free fraction is active
- Metabolized in liver: S-warfarin by CYP2C9 (key)
- Onset delayed (requires depletion of existing functional clotting factors): antithrombotic effect depends on reduction of factor X (t½ 24 h) and prothrombin (t½ 72 h)
- Therefore, bridge therapy with heparin/LMWH for at least 5 days at initiation
Monitoring: INR (prothrombin time ratio); therapeutic range typically INR 2–3 (2.5–3.5 for mechanical heart valves)
Genetic polymorphisms:
- CYP2C9*2, 2C93: Reduced enzyme activity → lower warfarin dose requirement (~25% Caucasians have ≥1 variant allele)
- VKORC1 variants: Affect enzyme susceptibility to warfarin inhibition → influence dose requirements
Drug Interactions (extensive):
Drugs that ↑ anticoagulant effect (↑ INR):
- CYP2C9 inhibitors: Fluconazole, amiodarone, metronidazole, trimethoprim
- Displacement from albumin: NSAIDs
- ↑ warfarin levels: Simvastatin, rosuvastatin
- Reduced vitamin K: Antibiotics (reduce gut flora), poor nutrition
Drugs that ↓ anticoagulant effect (↓ INR):
- CYP inducers: Rifampicin, carbamazepine, phenytoin, St. John's Wort
- ↑ vitamin K: Green leafy vegetables, vitamin K supplements
Reversal:
- Vitamin K (phytonadione) — oral or IV (onset 12–24 h)
- Fresh frozen plasma (FFP) — immediate but temporary
- Prothrombin complex concentrate (PCC) — faster and more complete than FFP
Indications:
- Atrial fibrillation (non-valvular and valvular)
- Mechanical prosthetic heart valves
- VTE treatment and prophylaxis
- Antiphospholipid syndrome
6. Direct Oral Anticoagulants (DOACs)
DOACs revolutionized anticoagulation by providing predictable pharmacokinetics, fixed dosing, and fewer drug/food interactions than warfarin.
Comparison Table (Goldman-Cecil Medicine)
| Property | Warfarin | Dabigatran | Rivaroxaban | Apixaban | Edoxaban |
|---|---|---|---|---|---|
| Mechanism | Vitamin K antagonist | Direct thrombin inhibitor | Factor Xa inhibitor | Factor Xa inhibitor | Factor Xa inhibitor |
| Target | Factors II, VII, IX, X, Proteins C&S | Factor IIa (thrombin) | Factor Xa | Factor Xa | Factor Xa |
| Administration | Once daily | Twice daily | Once or twice daily | Twice daily | Once daily |
| Monitoring | Regular INR required | Not routine | Not routine | Not routine | Not routine |
| Renal elimination | Minimal | ~80% | ~33% | ~27% | ~50% |
| Reversal agent | Vitamin K, PCC, FFP | Idarucizumab | Andexanet alfa | Andexanet alfa | Andexanet alfa |
a) Dabigatran (Direct Thrombin Inhibitor)
Mechanism: Direct, competitive inhibitor of both free and clot-bound thrombin (factor IIa); AT-independent
Pharmacokinetics:
- Prodrug (dabigatran etexilate) converted to active form by plasma and hepatic esterases
- Oral bioavailability ~6–7%
- t½: 12–17 h
- ~80% renal elimination → contraindicated in severe renal impairment (CrCl <30 mL/min)
- Minimal CYP interactions; substrate of P-glycoprotein
Monitoring: Not routinely required; dilute thrombin time or ecarin clotting time if needed
Reversal: Idarucizumab (humanized antibody fragment binding dabigatran with very high affinity)
Indications: Non-valvular atrial fibrillation, DVT/PE treatment and prevention
b) Rivaroxaban (Factor Xa Inhibitor)
Mechanism: Selective, direct inhibitor of factor Xa (both free and clot-bound); no need for AT cofactor
Pharmacokinetics:
- Oral bioavailability: ~80% (≥15 mg doses with food)
- t½: 5–9 h (young); 11–13 h (elderly)
- ~33% renal excretion; ~66% fecal
- CYP3A4 and P-gp substrate
Indications: Non-valvular AF, DVT/PE (acute treatment and secondary prevention), VTE prophylaxis post-surgery
c) Apixaban (Factor Xa Inhibitor)
Mechanism: Selective, reversible direct inhibitor of factor Xa
Pharmacokinetics:
- Oral bioavailability: ~50%
- t½: ~12 h
- ~27% renal excretion; primarily fecal elimination → favored in renal impairment
- CYP3A4 and P-gp substrate
Indications: Non-valvular AF, DVT/PE, VTE prophylaxis
d) Edoxaban (Factor Xa Inhibitor)
Mechanism: Selective, direct inhibitor of factor Xa
Pharmacokinetics:
- Oral bioavailability: ~62%
- t½: 10–14 h
- ~50% renal excretion
7. Reversal of Anticoagulation Summary
| Anticoagulant | Reversal Agent |
|---|---|
| UFH | Protamine sulfate (1 mg per 100 U) |
| LMWH | Protamine sulfate (partial) |
| Fondaparinux | No specific antidote (rFVIIa) |
| Warfarin | Vitamin K + PCC/FFP |
| Dabigatran | Idarucizumab |
| Rivaroxaban / Apixaban / Edoxaban | Andexanet alfa |
8. Heparin-Induced Thrombocytopenia (HIT)
A critical immune adverse effect:
- Type I HIT: Non-immune, mild, transient thrombocytopenia (first 2 days); not clinically significant
- Type II HIT: Immune-mediated; IgG antibodies against PF4-heparin complexes → platelet activation → paradoxical thrombosis despite thrombocytopenia
- Occurs day 5–14 of heparin exposure
- Thrombocytopenia: platelet count falls >50% from baseline
- Diagnose: 4T score + ELISA/functional assay
- Management: Stop ALL heparin; switch to argatroban (hepatic metabolism, preferred in renal failure) or bivalirudin; do NOT use LMWH (cross-reactive antibodies)
Key Comparison: UFH vs LMWH vs Fondaparinux
| Feature | UFH | LMWH | Fondaparinux |
|---|---|---|---|
| MW range | 3,000–30,000 Da | 4,000–8,000 Da | 1,728 Da (synthetic) |
| Anti-Xa : Anti-IIa ratio | 1:1 | 2:1 – 4:1 | Xa only (∞) |
| SC bioavailability | Poor | ~90% | ~100% |
| Monitoring | Required (aPTT) | Usually not needed | Usually not needed |
| HIT risk | 1–3% | ~0.1–0.5% | Negligible |
| Protamine reversal | Complete | Partial | None |
| Safe in pregnancy | Yes (does not cross placenta) | Yes | Avoid (limited data) |
| Renal caution | Less critical | Yes (CrCl<30) | Contraindicated (CrCl<30) |
Sources:
- Goodman & Gilman's The Pharmacological Basis of Therapeutics (Chapter 37 — Lipid Disorders; Chapter 5 — Drug Transport)
- Harrison's Principles of Internal Medicine 22E (Chapters on Oral Anticoagulants, Drug-Induced Liver Injury)
- Goldman-Cecil Medicine International Edition (Chapter 70 — Anticoagulant Therapy)
- Washington Manual of Medical Therapeutics (Lipid Management)
- Lippincott Illustrated Reviews: Pharmacology (Chapter 14 — Lipid-Lowering Drugs)
This is a shared conversation. Sign in to Orris to start your own chat.