Essay from General Medicine Topic: Nephrotic Syndrome
Reading File
Finding Sources
Searching PubMed
"nephrotic syndrome" management
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
I now have all the material needed. Let me write the comprehensive essay.
Nephrotic Syndrome: A Comprehensive Essay
Definition and Diagnostic Criteria
Nephrotic syndrome is a clinicopathological complex defined by massive proteinuria exceeding 3.5 g per 24 hours, serum albumin below 3.5 g/dL, generalized edema, and hyperlipidemia with lipiduria. In children, the equivalent threshold is proteinuria greater than 40 mg/m²/h or a urinary protein-to-creatinine ratio greater than 2.0 mg/mg. The syndrome reflects a fundamental derangement in glomerular capillary wall permeability, allowing plasma proteins to escape into the urine at a rate that exceeds the liver's compensatory synthetic capacity.
It is the third most common cause of end-stage kidney disease worldwide. Glomerular disease has diverse presentations and its workup requires serum chemistry, serology, urinalysis with microscopy, and quantification of proteinuria.
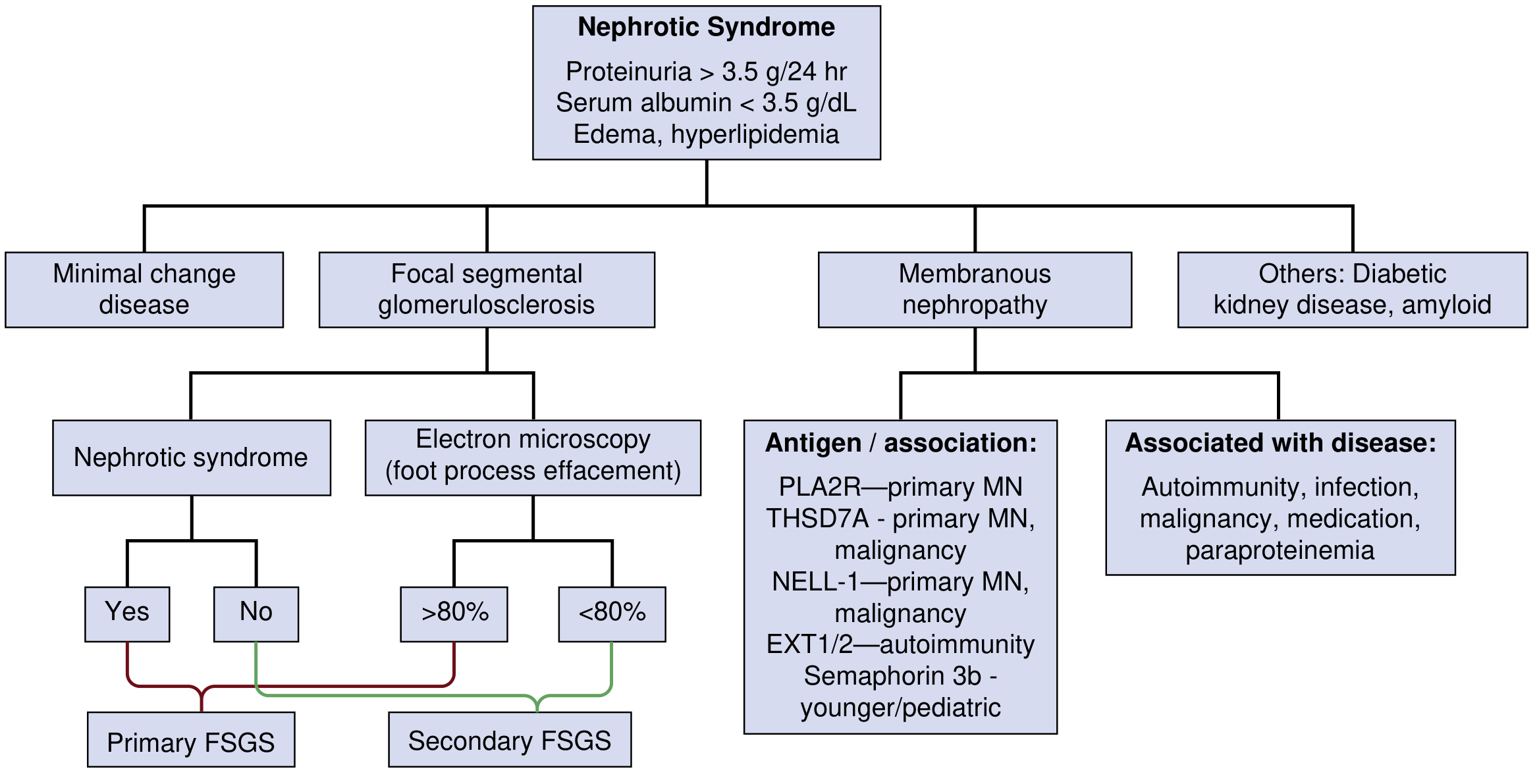
Fig. 1 - Schematic showing the most common etiologies of nephrotic syndrome, including classification of FSGS and membranous nephropathy antigens. (National Kidneys Foundation Primer on Kidney Diseases, 8e)
Pathophysiology
The glomerular capillary wall serves as both a size and charge barrier through which the plasma filtrate passes. It is composed of three layers: the fenestrated endothelium, the glomerular basement membrane (GBM), and the podocyte layer with its elaborate foot processes bridged by slit diaphragms. In nephrotic syndrome, either structural or physicochemical injury to this filtration barrier permits proteins - primarily albumin - to escape into the urinary space.
The cascade of events unfolds as follows:
-
Massive proteinuria - Increased glomerular permeability leads to urinary protein loss exceeding 3.5 g/day. The degree of selectivity matters: highly selective proteinuria consists mostly of low-molecular-weight proteins (albumin, transferrin), whereas poorly selective proteinuria also includes higher-molecular-weight globulins.
-
Hypoalbuminemia - Heavy proteinuria depletes serum albumin beyond the liver's compensatory synthetic rate. Increased renal catabolism of filtered albumin compounds this effect.
-
Edema - The fall in plasma oncotic pressure from hypoalbuminemia increases the flux of fluid into the interstitial spaces ("underfill" mechanism). Simultaneously, a primary renal salt retention can lead to "overfill" edema. Sodium retention occurs through several mechanisms: activation of the renin-angiotensin-aldosterone system (RAAS) driven by perceived hypovolemia, sympathetic nervous system stimulation, and reduced secretion of natriuretic peptides. Edema is characteristically soft, pitting, and most marked in the periorbital regions and dependent portions of the body. Severe hypoalbuminemia can produce pleural effusions and ascites.
-
Hyperlipidemia and lipiduria - Most patients develop elevated cholesterol, triglycerides, VLDL, LDL, and lipoprotein(a), with decreased HDL in some cases. This reflects a combination of increased hepatic lipoprotein synthesis (a compensatory response to low oncotic pressure), abnormal transport of circulating lipid particles, and decreased lipid catabolism. Lipiduria follows as lipoproteins also leak across the damaged glomerular wall, appearing in urine as free fat or oval fat bodies.
-
Hypercoagulability - The urinary loss of natural anticoagulants - protein C, protein S, plasminogen, and antithrombin III - creates a prothrombotic state. Renal vein thrombosis can occur when proteinuria exceeds 10 g/24 hr with severe hypoalbuminemia below 2 g/dL.
-
Susceptibility to infection - Loss of immunoglobulins in the urine predisposes to staphylococcal and pneumococcal infections. Spontaneous bacterial peritonitis occurs in 2-6% of children with nephrotic syndrome.
-
Additional deficiencies - Loss of transferrin causes iron deficiency anemia; loss of vitamin D-binding protein causes vitamin D deficiency; loss of thyroid-binding globulin affects thyroid hormone levels.
Aetiology and Classification
Nephrotic syndrome is broadly divided into primary (idiopathic) causes arising from intrinsic glomerular disease, and secondary causes resulting from systemic disorders.
Primary (Idiopathic) Causes
The three most common primary glomerular diseases manifesting as nephrotic syndrome share the pathological hallmark of extensive podocyte foot process effacement on electron microscopy:
1. Minimal Change Disease (MCD)
2. Focal Segmental Glomerulosclerosis (FSGS)
3. Membranous Nephropathy (MN)
Secondary Causes
- Diabetic kidney disease (DKD) - the most common cause of kidney failure in the United States
- Amyloidosis
- Lupus nephritis (SLE)
- Infections - hepatitis B, hepatitis C, HIV
- Drugs - NSAIDs, gold, penicillamine, heroin
- Malignancies - Hodgkin lymphoma (MCD), solid tumors (MN)
- Monoclonal immunoglobulin-associated disease
- Hereditary/genetic causes - mutations in podocyte genes
Major Causes of Primary Nephrotic Syndrome
1. Minimal Change Disease (MCD)
MCD is the most common cause of nephrotic syndrome in children, accounting for 70-90% of pediatric cases, but only ~15% of adult nephrotic syndrome. The age of peak incidence is 1 to 7 years.
Pathogenesis: The leading hypothesis implicates a T-cell-mediated mechanism - circulating molecules injure podocytes causing proteinuria. Evidence includes: the clinical response to corticosteroids, associations with Hodgkin lymphoma, and the observation that T-cell hybridomas from affected patients secrete a factor provoking proteinuria in rats. Candidate molecules include interleukin-13 (IL-13) and angiopoietin-like-4. More recently, a subset of patients has been found to have anti-nephrin antibodies, and circulating endothelial biomarkers suggest MCD is not purely a "podocytopathy." Urinary CD80 (B7.1) levels correlate with disease activity.
Morphology:
- Light microscopy (LM): Glomeruli appear entirely normal (hence the name)
- Immunofluorescence (IF): No immune deposits
- Electron microscopy (EM): Diffuse effacement of podocyte foot processes (the pathognomonic finding), along with vacuolization, microvillus formation, and focal detachments
The proximal convoluted tubular cells are heavily laden with protein droplets and lipids due to tubular reabsorption of leaked molecules.
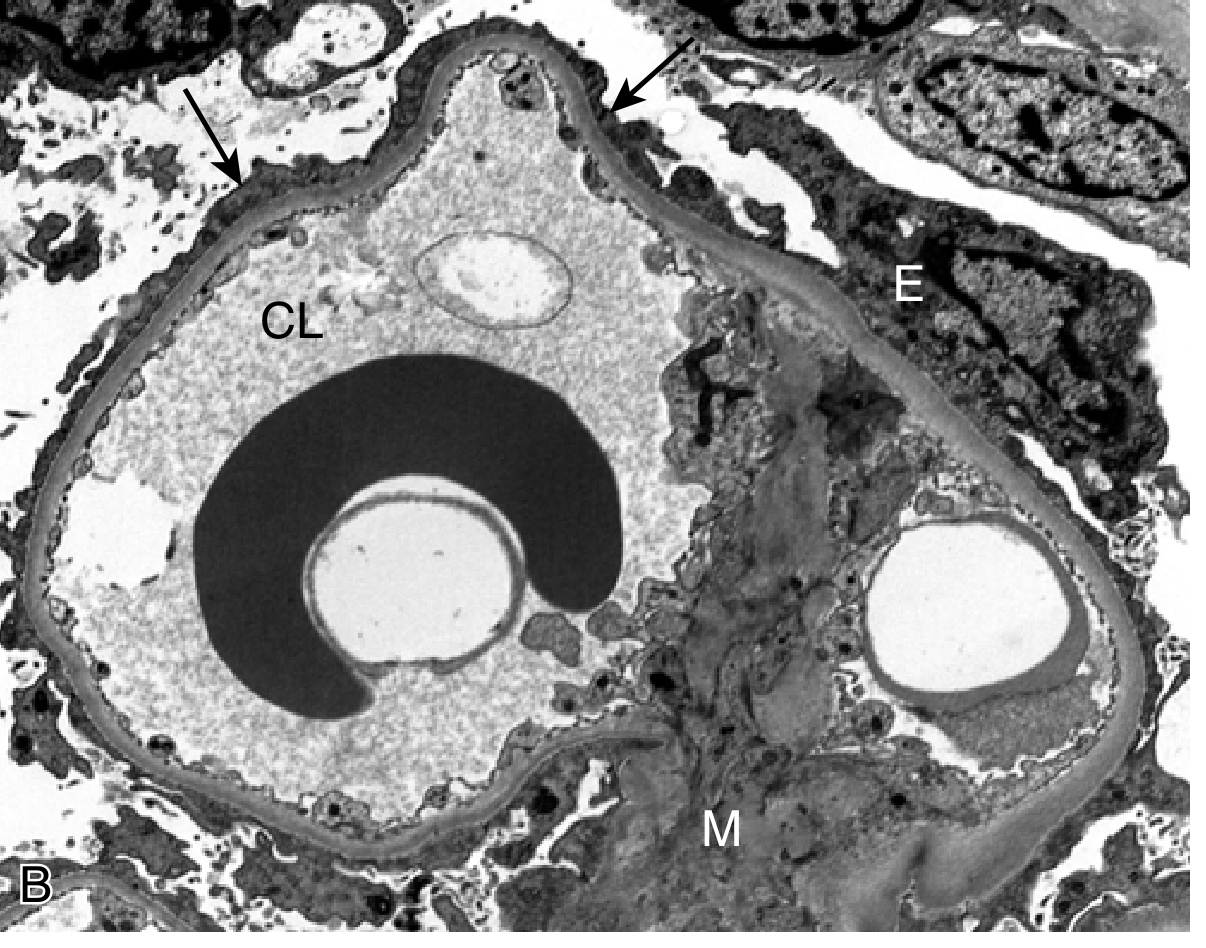
Fig. 2 - Electron micrograph of Minimal Change Disease showing diffuse effacement of podocyte foot processes (arrows). CL = capillary lumen; E = epithelial cell; M = mesangium. (Robbins & Kumar Basic Pathology)
Clinical features:
- Abrupt onset of nephrotic syndrome in an otherwise healthy child
- Selective proteinuria (mainly albumin)
- Renal function usually preserved
- Hematuria and hypertension are not typical
- In children without atypical features, kidney biopsy is NOT required before treatment
Treatment and prognosis:
- Mainstay: corticosteroids (prednisone 60 mg/m²/day or 2 mg/kg/day)
- Over 90% of children respond; response is defined as resolution of proteinuria within 8 weeks
- Proteinuria recurs in more than two-thirds of initial responders
- Steroid-sparing agents for frequent relapsers: cyclophosphamide, mycophenolate mofetil (MMF), rituximab (anti-CD20), calcineurin inhibitors (CNIs)
- Frequently-relapsing NS (FRNS): ≥2 relapses within 6 months or ≥4 relapses within 12 months
- Steroid-dependent NS (SDNS): Two consecutive relapses during steroid therapy, or inability to maintain remission without continuous steroids
2. Focal Segmental Glomerulosclerosis (FSGS)
FSGS is a histopathological pattern of glomerular scarring due to podocyte injury. It contributes approximately 25% of adult nephrotic syndrome and is the most common cause of idiopathic nephrotic syndrome among those of African descent. It accounts for <15% of pediatric nephrotic syndrome.
Classification:
FSGS is classified into primary, secondary, and genetic forms:
- Primary FSGS: Due to an undiscovered circulating podocyte permeability factor; typically presents with acute onset of nephrotic syndrome, low albumin, and proteinuria >3.5 g/day
- Secondary FSGS:
- Viral: HIV-associated nephropathy, parvovirus B19, CMV, COVID-19
- Drug-induced: Heroin, interferons, pamidronate, sirolimus, calcineurin inhibitors, anabolic steroids
- Adaptive: Reduced nephron mass, obesity-related glomerulopathy, low birth weight, unilateral renal agenesis, sickle cell anemia
- Genetic FSGS: Mutations in podocyte genes including NPHS1 (nephrin), NPHS2 (podocin), PLCE1, INF2, ACTN4, and APOL1 (strongly associated with FSGS in African descent)
Morphology:
- LM: Focal (affecting only some glomeruli) and segmental (involving a portion of the glomerular tuft) sclerosis. Five variants: cellular, tip, perihilar, collapsing, and NOS
- EM: Varying degrees of foot process effacement; >80% effacement favors primary FSGS; <80% favors secondary
- Collapsing variant: Strongly associated with viral etiologies (HIV, COVID-19); presents with abrupt onset of severe nephrotic syndrome and rapidly progressive loss of kidney function
Clinical features: In addition to nephrotic-range proteinuria, patients with FSGS may also have concomitant hypertension, reduced GFR, and microscopic hematuria - features that distinguish it from MCD.
Prognosis and treatment:
- Immunosuppression (corticosteroids, calcineurin inhibitors, mycophenolate mofetil) for primary FSGS
- Secondary FSGS: address underlying cause; avoid unnecessary immunosuppression
- Primary FSGS recurs rapidly after kidney transplant (supporting the circulating factor hypothesis)
- Persistent hematuria or poor response to immunosuppression indicates progression toward ESKD
- The APOL1 gene polymorphism in patients of African descent confers increased risk of FSGS and advanced kidney disease
3. Membranous Nephropathy (MN)
MN is the most common cause of adult nephrotic syndrome in whites, typically presenting in the fourth to fifth decade of life, with a 2:1 male-to-female ratio.
Pathogenesis: MN has the best-characterized pathogenesis among the nephrotic syndrome causes:
- Anti-PLA2R antibodies (anti-phospholipase A2 receptor): present in ~70% of primary MN; a critical diagnostic and monitoring biomarker
- Anti-THSD7A antibodies (thrombospondin type-1 domain containing 7A): present in 1-3% of primary MN, also associated with malignancy
- Newer antigens: NELL-1, EXT1/2 (associated with autoimmune disease), Semaphorin 3B (associated with pediatric cases)
- Secondary causes: SLE (EXT1/2), hepatitis B infection, NSAIDs, solid tumors, paraproteinemias
Morphology:
- LM: Normal glomerular architecture initially; methenamine silver stain shows subepithelial spikes along capillary walls in later stages
- IF: Granular deposition of IgG and C3 along capillary walls (characteristic granular pattern)
- EM: Subepithelial and intramembranous immune complex deposits - the hallmark; plus foot process effacement shared with MCD and FSGS
- Biopsies can now be stained for PLA2R, THSD7A, EXT1/2, NELL-1, and Sema3B antigens
Natural history:
The "rule of thirds" applies:
- One-third undergo spontaneous remission
- One-third achieve partial remission
- One-third progress (persistent nephrotic syndrome or declining GFR)
Treatment:
- Indications for immunosuppression: persistent nephrotic syndrome and/or declining kidney function
- Options: Rituximab (increasingly first-line), calcineurin inhibitors, cyclophosphamide + steroids
- PLA2R antibody titers guide response to immunosuppressive therapy
- If progressive course: evaluate for superimposed crescentic GN or acute interstitial nephritis
Clinical Features and Complications
Core Features
| Feature | Mechanism |
|---|---|
| Massive proteinuria (>3.5 g/day) | Increased GBM permeability |
| Hypoalbuminemia (<3.5 g/dL) | Urinary protein loss + increased renal catabolism |
| Generalized edema | Decreased oncotic pressure + sodium retention |
| Hyperlipidemia | Increased hepatic lipoprotein synthesis + decreased catabolism |
| Lipiduria (oval fat bodies, fatty casts) | Lipoprotein leakage across damaged GBM |
Complications
Thromboembolism: Loss of antithrombin III, protein C, protein S, and plasminogen from the urine creates hypercoagulability. Renal vein thrombosis, deep vein thrombosis, and pulmonary embolism can occur. This complication is most prominent in MN and amyloidosis.
Infections: Particularly staphylococcal and pneumococcal infections due to urinary loss of immunoglobulins. Spontaneous bacterial peritonitis is a recognized complication in children (2-6%).
Acute kidney injury (AKI): Due to low effective circulating volume from hypoalbuminemia and reduced oncotic pressure.
Nutritional and metabolic deficiencies:
- Iron deficiency anemia (transferrin loss)
- Vitamin D deficiency (vitamin D-binding protein loss)
- Hypothyroidism (thyroid-binding globulin loss)
- Hypogammaglobulinemia predisposing to sepsis
Cardiovascular disease: Chronic hyperlipidemia accelerates atherosclerosis.
Drug toxicity: Hypoalbuminemia alters the pharmacokinetics of protein-bound drugs, increasing the free fraction and toxicity risk.
Investigation
Urine:
- 24-hour urinary protein (>3.5 g/day) or spot urine protein-to-creatinine ratio (>2-3.5 mg/mg)
- Urinalysis: heavy proteinuria, lipiduria, oval fat bodies, fatty casts, maltese crosses under polarized light
- Urinary microscopy: absence of red cell casts (distinguishes from nephritic syndrome)
Blood:
- Serum albumin (decreased)
- Lipid profile: total cholesterol, LDL, triglycerides (elevated), HDL (may be decreased)
- Renal function: creatinine, urea, eGFR
- Complement levels (C3, C4) - may be low in secondary causes (SLE, MPGN)
- ANCA, ANA, anti-dsDNA (for secondary causes)
- Anti-PLA2R and anti-THSD7A antibodies (for membranous nephropathy)
- Serum protein electrophoresis, free light chains (to exclude paraproteinemia)
- HBsAg, anti-HCV, HIV serology
- Blood glucose, HbA1c (for diabetic nephropathy)
Kidney biopsy:
- Indicated in adults with nephrotic syndrome (essential for diagnosis)
- In children with typical presentation (age 1-8 years, no hematuria, normal complement, no hypertension), empirical steroid therapy is started without biopsy
- Atypical features warranting biopsy in children: age <1 year, macroscopic hematuria, hypertension, hypocomplementemia, extrarenal symptoms (rash, arthritis)
Management
General (Non-Disease-Specific) Measures
1. Dietary modifications:
- Low-salt diet to reduce edema
- Protein restriction: 0.8 to 1 g/kg/day (excessive protein intake increases proteinuria without benefit)
- Hypercaloric diet in severe disease
2. Antiproteinuric therapy:
- ACE inhibitors (ACEi) or angiotensin receptor blockers (ARBs) reduce intraglomerular pressure and proteinuria; considered cornerstone therapy regardless of etiology
- Adding a loop diuretic to ACEi/ARB reduces proteinuria further but may increase serum creatinine
3. Edema management:
- Loop diuretics (furosemide) - first-line; note that hypoalbuminemia reduces protein-binding of furosemide, increasing its volume of distribution
- Intravenous albumin + loop diuretic combination is used in severe diuretic-resistant cases (though iso-oncotic albumin infusion alone does not reliably induce negative sodium balance)
- Thiazide diuretics added as adjuncts
4. Hyperlipidemia:
- Statin therapy for dyslipidemia
5. Anticoagulation:
- Prophylactic anticoagulation considered for patients at high risk of thrombosis (severe proteinuria >10 g/day, serum albumin <2 g/dL), particularly in membranous nephropathy
6. Infection prevention:
- Pneumococcal and influenza vaccination
- Prompt treatment of infections
Disease-Specific Treatment (Summary)
| Cause | First-Line Treatment | Second-Line / Steroid-Sparing |
|---|---|---|
| MCD (children) | Prednisolone 2 mg/kg/day (8 weeks) | Cyclophosphamide, MMF, Rituximab, CNIs |
| MCD (adults) | Prednisolone | CNIs, Rituximab |
| Primary FSGS | Prednisolone | CNIs, MMF, Rituximab |
| Primary MN | Observe (one-third remit spontaneously) | Rituximab (first-line), CNIs, Cyclophosphamide + steroids |
| Secondary causes | Treat underlying cause | ACEi/ARB antiproteinuric therapy |
| DKD | Glycemic control, BP control, ACEi/ARB, SGLT2i |
Nephrotic vs. Nephritic Syndrome: Key Distinction
| Feature | Nephrotic | Nephritic |
|---|---|---|
| Proteinuria | Massive (>3.5 g/day) | Mild-moderate |
| Haematuria | Absent or microscopic | Present (often macroscopic) |
| Red cell casts | Absent | Characteristic |
| Oedema | Marked | Mild |
| Hypertension | Variable | Prominent |
| GFR | Usually preserved initially | Often reduced |
| Hypoalbuminaemia | Severe | Mild |
Special Populations
Children
In children aged 1-8 years with a typical presentation of nephrotic syndrome (no hematuria, normal complement, normal BP), empirical steroid therapy is started without biopsy. MCNS is the assumed diagnosis. Congenital nephrotic syndrome (Finnish type, due to NPHS1 mutations) presents within the first 3 months of life and requires early nephrectomy and dialysis/transplant.
Adults
In adults, biopsy is mandatory as MCD, FSGS, MN, and secondary causes are equally possible and require different treatments.
Pregnancy
Nephrotic syndrome in pregnancy raises the possibility of pre-eclampsia as well as primary glomerular disease; careful evaluation is required.
Prognosis
Prognosis varies markedly by underlying cause:
- MCD in children: Excellent prognosis; >90% initial steroid response; most achieve long-term remission though relapses are common
- Primary FSGS: Variable; patients with non-nephrotic proteinuria have the best outcomes; persistent nephrotic syndrome or hematuria and poor steroid response predict progression to ESKD
- Primary MN: Rule of thirds applies; anti-PLA2R titer is a reliable guide
- Secondary causes: Prognosis determined by the underlying systemic disease
- Complications (thromboembolism, infection, AKI) worsen overall outcomes
References and Sources
- National Kidneys Foundation Primer on Kidney Diseases, 8th Edition - Chapters 16, 18, 19, 20
- Robbins, Cotran & Kumar Pathologic Basis of Disease - Chapter on Glomerular Diseases
- Robbins & Kumar Basic Pathology - Chapter 12
- Comprehensive Clinical Nephrology, 7th Edition - Chapter on Minimal Change Disease
- Brenner and Rector's The Kidney, 2-Volume Set - Chapter 50 (Edema in Nephrotic Syndrome)
- Campbell Walsh Wein Urology, 3-Volume Set - Pediatric Nephrotic Syndrome
Recent Evidence Note: A 2025 Cochrane systematic review (PMID 40337980) on interventions for idiopathic steroid-resistant nephrotic syndrome in children updates treatment considerations for this subgroup. A 2026 meta-analysis (PMID 41843217) specifically reports rituximab-induced hypogammaglobulinemia as a significant complication in childhood nephrotic syndrome - clinicians should monitor immunoglobulin levels when using rituximab.
This is a shared conversation. Sign in to Orris to start your own chat.