Sickle cell disease
Reading File
Finding Sources
Searching PubMed
"sickle cell disease"[MeSH Terms] AND treatment
Finding Sources
Searching PubMed
"sickle cell disease" AND "hydroxyurea OR gene therapy OR voxelotor"
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive information. Let me compile the full answer.
Sickle Cell Disease (SCD)
Genetics and Epidemiology
Sickle cell disease is an autosomal recessive hemoglobinopathy caused by a single point mutation in the β-globin gene on chromosome 11 - an adenine-to-thymine transversion at codon 6 (A6T), which substitutes valine for glutamic acid at the 6th position of the β-globin chain. This creates sickle hemoglobin (HbS).
- Approximately 250,000 children worldwide are born with homozygous SCD annually
- In the United States, about 8% of people of African descent are heterozygous carriers (sickle cell trait), and about 1 in 600 have sickle cell anemia
- The HbS allele is prevalent in areas where Plasmodium falciparum malaria was endemic (equatorial Africa, parts of India, southern Europe, Middle East) because heterozygosity confers protection against malaria
- Median age at death: 48-58.5 years in women, 42-53 years in men
Genotypic variants:
| Genotype | Disease Severity |
|---|---|
| HbSS (homozygous) | Most severe |
| HbSβ⁰-thalassemia | Severe (similar to HbSS) |
| HbSC | Moderate |
| HbSβ⁺-thalassemia | Mild-moderate |
| HbAS (sickle trait) | Usually asymptomatic |
Pathophysiology
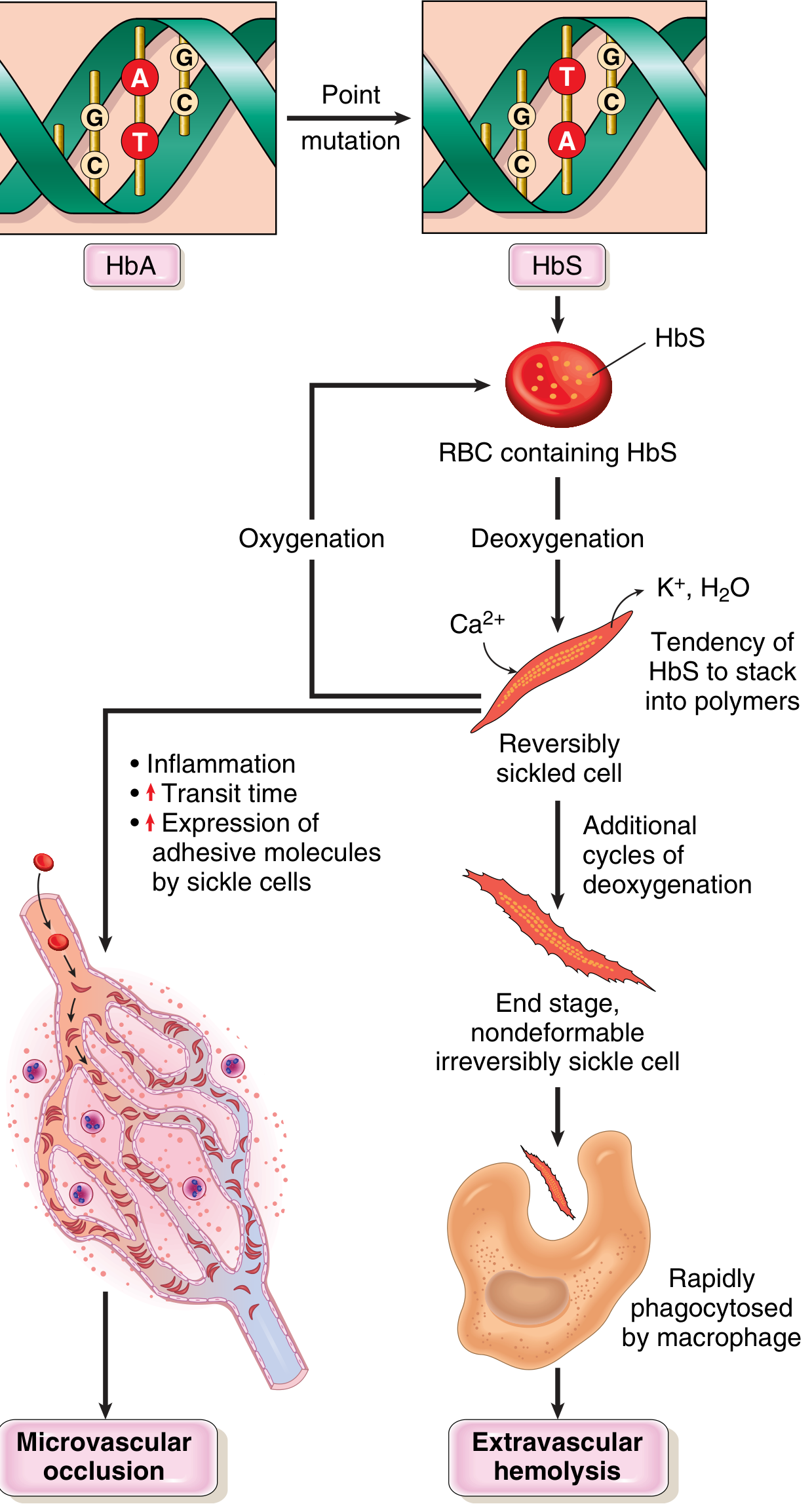
Pathophysiology of sickle cell disease (Robbins & Kumar Basic Pathology)
The core mechanism involves HbS polymerization on deoxygenation:
-
Polymer formation - Deoxygenated HbS molecules change conformation and self-associate into long polymers via contacts involving the abnormal valine residue. These distort red cells into the characteristic sickle/crescent shape.
-
Reversible vs. irreversible sickling - Early sickling episodes are reversible on reoxygenation in the lungs. However, each cycle causes calcium influx, loss of K⁺ and water, and membrane skeleton damage. Repeated cycles create irreversibly sickled cells (ISCs) prone to hemolysis. Mean red cell lifespan is reduced to ~20 days (vs. 120 days normal).
-
Three major determinants of sickling severity:
- Intracellular HbS concentration - Higher concentration worsens sickling. HbF inhibits polymerization (explains why neonates are protected until HbF falls at 5-6 months)
- HbS percentage - HbA (in carriers) and HbF interpose and retard polymerization
- Transit time through microvasculature - Tissues with sluggish flow (spleen, bone marrow) are most susceptible; inflammation prolongs transit times
-
Dual consequences:
- Hemolytic anemia - Extravascular (macrophage phagocytosis of ISCs) and intravascular hemolysis; releases free hemoglobin, which scavenges nitric oxide (NO) and releases arginase (destroys arginine, the substrate for NO synthesis)
- Vaso-occlusion - Sickled cells adhere to endothelium; triggered by infection, inflammation, dehydration, and acidosis. Produces ischemia-reperfusion injury, inflammatory, thrombotic, and oxidant stress
-
NO deficiency - A state of endothelial dysfunction results, skewing the vascular balance toward vasoconstriction, platelet activation, and upregulation of cell adhesion molecules. Heme released post-hemolysis also activates TLR4 and inflammasome signaling, amplifying inflammation.
Clinical Manifestations
Acute Complications
1. Vaso-occlusive (Pain) Crises
- The hallmark of SCD; most common cause of hospitalization
- Triggered by infection, dehydration, stress, cold, or unprovoked
- Infants: dactylitis (hand-foot syndrome - painful swelling of fingers/toes)
- Older patients: arms, legs, trunk, back pain
- Almost 30% of adults experience pain on >95% of days
- Diagnosis is clinical; no specific diagnostic test exists
2. Acute Chest Syndrome (ACS)
- A lung injury syndrome analogous to ARDS
- Defined by: new pulmonary opacity on CXR (≥1 complete segment, consolidation not atelectasis) + chest pain, fever, tachypnea, or hypoxia
- Typically begins 24-72 hours after onset of a pain crisis
- Most common cause of mortality in adults (accounts for 28% of deaths in long-term studies)
- Causes: infection, fat embolism from infarcted bone marrow, in situ sickling
3. Stroke
- Occurs in ~11% of children before age 20
- Most common in young children and older adults
- Transcranial Doppler (TCD) screening identifies children at high risk; elevated velocities prompt primary prevention with chronic transfusion
4. Aplastic Crisis
- Transient cessation of red cell production, usually triggered by Parvovirus B19 infection
- Dangerous given already short red cell lifespan
5. Splenic Sequestration
- Acute pooling of blood in the spleen (especially in children <5 years)
- Presents with rapid splenomegaly, falling hemoglobin, circulatory compromise
6. Infections
- Functional asplenia (autosplenectomy) by adulthood leads to high susceptibility to encapsulated organisms: Streptococcus pneumoniae, Haemophilus influenzae, Neisseria meningitidis
- Salmonella osteomyelitis is characteristic
Chronic Complications
| Organ System | Manifestation |
|---|---|
| Bone | Avascular necrosis (femoral/humoral head), "crewcut" skull X-ray, Salmonella osteomyelitis |
| Spleen | Autosplenectomy (repeated infarcts) → functional asplenia |
| Kidney | Hyposthenuria, microalbuminuria, papillary necrosis, end-stage renal disease |
| Lung | Pulmonary hypertension, chronic lung disease |
| Eye | Proliferative retinopathy, vitreous hemorrhage |
| CNS | Stroke, cognitive impairment, silent cerebral infarcts |
| Heart | Cardiomegaly, high-output failure |
| Liver | Pigment gallstones (bilirubin), hepatic sequestration |
| Skin | Leg ulcers (ankle) |
| GU | Priapism → penile fibrosis, erectile dysfunction; enuresis |
| Growth | Delayed puberty, growth retardation |
Diagnosis
- Newborn screening (mandatory in the US): Hemoglobin electrophoresis or HPLC - pattern HbSS shows only HbS and HbF (no HbA)
- Peripheral blood smear: Elongated, crescent/spindle-shaped irreversibly sickled cells, target cells, Howell-Jolly bodies (asplenia)
- Sickling tests (sodium metabisulfite or solubility) - confirm HbS presence but do NOT differentiate HbAS from HbSS
- Hemoglobin electrophoresis or HPLC: Gold standard for genotype
- Tandem mass spectrometry: Identifies the specific amino acid change
- DNA sequencing: Identifies the specific globin gene mutation
Management
General Measures
- High fluid intake and good nutrition
- Folic acid supplementation (accelerated RBC turnover depletes folate)
- Education about triggers and "red flag" symptoms requiring emergency care
- Regular outpatient monitoring for chronic complications
- Vaccinations: pneumococcal, Haemophilus, meningococcal, influenza
- Penicillin prophylaxis in children (until age 5) against pneumococcal infection
- Psychological and social support
Red Flag Symptoms Requiring Hospital Admission
- Severe pain unresponsive to usual analgesia
- Fever >38°C
- Chest pain or hypoxia
- Neurological signs
- Acute splenic enlargement
- Priapism >4 hours
Acute Pain Management
- Mild: NSAIDs, acetaminophen, oral fluids, rest at home
- Severe: Hospital admission; parenteral opioids (patient-controlled analgesia reduces total opiate intake); monitor for respiratory depression
- Incentive spirometry to reduce ACS risk
- Taper opiates as pain resolves
Acute Chest Syndrome Treatment
- Oxygen, bronchodilators
- Empirical antibiotics (cover atypical organisms)
- Simple or exchange transfusion
- Incentive spirometry
Transfusion
- Acute indications: Aplastic crisis, acute chest syndrome, stroke, multiorgan failure, symptomatic severe anemia - NOT indicated for simple pain crises
- Chronic indications: Primary/secondary stroke prevention, recurrent pain or ACS not responding to hydroxyurea
- Exchange transfusion: Preferred for stroke and high-risk surgery (cardiac, neurosurgery); target HbS <30%
- Simple transfusion: For low-to-moderate risk surgery, target Hb >9 g/dL
- Goal: Reduce HbS% to <40%, maintain Hb ~10 g/dL
- Risks: Alloimmunization, iron overload, transfusion reactions
Disease-Modifying Therapies
1. Hydroxyurea (Hydroxycarbamide)
- Mechanism: Increases HbF levels (inhibits HbS polymerization); reduces cellular adhesion, hemolysis, and neutrophil count
- Dose: Up to 30 mg/kg/day (dose-escalation protocols superior to fixed 20 mg/kg/day)
- Benefits: Reduces pain crises, ACS, hospitalizations, stroke risk (when TCD elevated)
- Recommended for ALL infants, children, and adolescents with SCD regardless of severity; adults with recurrent pain, severe/recurrent ACS, or symptomatic anemia
- Side effects: Myelosuppression (requires regular CBC monitoring); avoid in pregnancy (potential teratogenicity and effects on spermatogenesis)
2. L-Glutamine
- Reduces oxidative stress in sickle red cells
- FDA-approved; reduces acute complications
3. Crizanlizumab
- Anti-P-selectin monoclonal antibody; blocks sickle cell adhesion to endothelium
- Reduces frequency of vaso-occlusive crises
4. Voxelotor (GBT440)
- Increases Hb-O₂ affinity; stabilizes oxygenated HbS, reducing polymerization
- Raises hemoglobin level and reduces hemolysis markers
5. Blood and Stem Cell Transplantation
- The only established curative therapy
- Allogeneic HSCT from HLA-matched sibling donor; best outcomes in young patients with severe disease before organ damage accumulates
- 5-year event-free survival >90% with matched sibling donor in children
- Limited by donor availability and transplant-related morbidity
6. Gene Therapy (Emerging)
- Betibeglogene autotemcel (Zynteglo) and lovotibeglogene autotemcel (Lyfgenia) - FDA-approved lentiviral gene addition therapies (2023)
- Exa-cel (Casgevy) - CRISPR-Cas9 gene editing (reactivates HbF by disrupting BCL11A); FDA-approved December 2023 - the first approved CRISPR therapy
- These approaches offer curative potential without donor dependency
Special Situations
Pregnancy
- 6-fold increased risk of maternal death vs. controls
- Complications: more frequent pain crises, ACS, VTE (1.7-10x more frequent), preeclampsia, preterm birth, fetal growth restriction
- Hydroxyurea is contraindicated (teratogenic); NSAIDs avoided after 30 weeks
- Regional anesthesia preferred for cesarean section
- Prophylactic transfusion may reduce complications; therapeutic transfusion goal: HbS <40%, Hb ~10 g/dL
- Low-dose aspirin (81 mg/day) for preeclampsia prevention
Surgery and Anesthesia
- General anesthesia increases risk of pain crises and ACS
- Preoperative hydration and oxygenation are essential
- Low-to-moderate risk surgery: simple transfusion to Hb >9 g/dL
- High-risk surgery (cardiac/neuro): preoperative exchange transfusion
Stroke Prevention
- Annual TCD screening in children aged 2-16 years
- If TCD velocity elevated (>200 cm/s): chronic transfusion program reduces stroke risk by ~90%
- Hydroxyurea is an alternative when transfusion is not available
Morphological Summary (Pathology)
- Peripheral smear: sickled cells, target cells, polychromasia, nucleated RBCs
- Bones: "crewcut" skull (marrow expansion), H-shaped vertebrae, avascular necrosis
- Spleen: autosplenectomy in adults (fibrous remnant); splenomegaly in young children
- Gallbladder: pigment gallstones (bilirubin)
- Heart/liver/kidney: fatty changes from hypoxia
- Bone marrow: erythroid hyperplasia; extramedullary hematopoiesis in liver/spleen
Sources:
- Goldman-Cecil Medicine, Ch. 149 (Sickle Cell Disease)
- Robbins & Kumar Basic Pathology, Ch. 10 (Sickle Cell Anemia)
- Murray & Nadel's Textbook of Respiratory Medicine (Acute Chest Syndrome)
- Rosen's Emergency Medicine (SCD in Pregnancy)
Recent literature note: A 2026 Lancet review on sickle cell disease (PMID 41831848) and a 2023 Annual Review of Genomics article on curative approaches (PMID 37624668) are the most current comprehensive overviews, which may include updated data on gene therapy approvals and long-term outcomes not yet in the textbooks.
This is a shared conversation. Sign in to Orris to start your own chat.