Achondroplasia
Reading File
Searching PubMed
achondroplasia FGFR3 treatment
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have a thorough, multi-source picture. Here is the complete synthesis:
Achondroplasia

Definition & Epidemiology
Achondroplasia is the most common skeletal dysplasia and the most common cause of disproportionate short stature (dwarfism). Incidence is approximately 1 in 15,000 to 1 in 40,000 live births, affecting all ancestries equally.
- Robbins & Kumar Basic Pathology, p. 3561
- Thompson & Thompson Genetics, p. 450
Genetics & Molecular Pathogenesis
| Feature | Detail |
|---|---|
| Gene | FGFR3 (fibroblast growth factor receptor 3) |
| Mutation | c.1138G>A (~98%) or c.1138G>C (1-2%), both causing p.Gly380Arg substitution |
| Effect | Gain-of-function - constitutive, ligand-independent activation of FGFR3 |
| Inheritance | Autosomal dominant |
| De novo rate | ~80% of cases are new mutations, exclusively from the paternal germline, increasing in frequency with advanced paternal age (>35 years) |
| Allele state | Almost all affected individuals are heterozygous |
Mechanism: FGFR3 normally inhibits chondrocyte proliferation in the growth plate to coordinate bone growth. In achondroplasia, the constitutively active receptor pathologically suppresses chondrocyte proliferation within the growth plate, leading to shortening of long bones (endochondral ossification is impaired). Membranous ossification is unaffected, so the skull vault grows normally while the cranial base (formed by endochondral bone) is small.
- Thompson & Thompson Genetics, p. 450 (Pathogenesis)
- Robbins & Kumar Basic Pathology, p. 3561
- Goldman-Cecil Medicine, p. 2663
Clinical Features
Skeletal
- Rhizomelic shortening - proximal limbs (humerus, femur) most affected
- Relatively normal trunk length
- Trident hand configuration (digits separated into three groups)
- Macrocephaly with frontal bossing (prominent forehead)
- Midface hypoplasia (flat nasal bridge, small cranial base)
- Lumbar lordosis - greatly exaggerated; spinal canal narrows from upper to lower lumbar segments
- Long bones appear disproportionately wide relative to length
- Radiograph: decreasing interpedicular distance from L1 to L5 (hallmark)
Neurological
- Foramen magnum stenosis in ~10-20% - can compress brainstem at craniocervical junction
- Untreated, critical compression may cause: central apnea, hypotonia, weakness, quadriparesis, failure to thrive, sudden death (up to 5% in first year of life)
- Lumbar spinal stenosis - worsens with age; symptomatic incidence 37-89%, usually presenting in 3rd-4th decade
- Hydrocephalus (less common)
Skull & ENT
- Small cranial foramina - midface hypoplasia causes dental crowding, obstructive sleep apnea, and chronic otitis media
- Small foramen magnum
Other
-
Normal intelligence
-
Delayed motor development (from hypotonia + hyperextensible joints + difficulty balancing large head)
-
Obesity tendency
-
Hypertension
-
Genu varum (bow legs)
-
Thompson & Thompson Genetics, p. 450-451
-
Goldman-Cecil Medicine, p. 2663
Spinal Complications (Key Orthopedic Points)
Thoracolumbar kyphosis:
- Present at birth in the majority; frequency is 87% at ages 1-2 years, 39% at 2-5 years, 11% at 5-10 years
- Usually resolves once walking begins
- Management: early bracing (TLSO) if kyphosis >30° on prone lateral radiograph; avoid unsupported sitting
- Surgery indications: documented progression, kyphosis >50°, or neurologic deficits; delayed until age 4 unless rapidly progressive
Lumbar stenosis:
-
Interpedicular distance decreases L1 to L5 (opposite of normal)
-
Pedicles are short (reduced AP canal diameter by ~40%)
-
Treatment: surgical decompression ± stabilization
-
Campbell's Operative Orthopaedics 15th Ed 2026, p. 2183-2184
Diagnosis
- Usually made clinically and radiographically at birth
- DNA testing for FGFR3 variants is confirmatory in ambiguous cases but generally not required
- Prenatal diagnosis possible by ultrasound (shortened femur length, macrocephaly) and/or molecular testing
Management
Surveillance (Lifelong, Age-Stratified)
| Age | Focus |
|---|---|
| Infancy | Polysomnography (sleep apnea), MRI of craniocervical junction, serial neurological exams |
| Early childhood | Otitis media, hearing, obstructive apnea, thoracolumbar kyphosis |
| Later childhood/adult | Spinal stenosis, genu varum, obesity, hypertension, dental crowding |
- Avoid activities risking craniocervical injury (contact/collision sports, trampolines, diving, vaulting in gymnastics)
- Craniocervical decompression surgery when brainstem compression confirmed
Drug Therapy - Vosoritide
Vosoritide (trade name Voxzogo) is a once-daily subcutaneous injection, a CNP (C-type natriuretic peptide) analogue that counteracts FGFR3 overactivation by stimulating the NPR-B receptor. It was approved by FDA and EMA for children with achondroplasia. The landmark Phase 3 RCT (Savarirayan et al., Lancet 2020) demonstrated significant increase in annualized growth velocity.
Three recent meta-analyses (2026) confirm its efficacy and acceptable safety profile in real-world settings (PMIDs: 42026358, 41934413, 41424367).
- Goldman-Cecil Medicine, p. 2664 (vosoritide as daily subcutaneous CNP analogue)
Growth Hormone & Limb Lengthening
Both remain controversial. Growth hormone can increase growth velocity but effects are modest and do not normalize height. Surgical limb lengthening (distraction osteogenesis) can add 20-30 cm but requires multiple surgeries and prolonged rehabilitation.
Psychosocial
Social adjustment support, adaptive equipment (driving, toileting), school/work accommodation advocacy, and peer support groups are important components of care.
Anesthetic Considerations
-
Airway difficulty: midface hypoplasia with pharynx small relative to tonsils/adenoids/tongue - prone to upper airway obstruction; mask seal difficult with flat nasal bridge
-
Avoid neck hyperextension - risk of foramen magnum compression
-
Prefer video laryngoscopy or fiberoptic intubation; use smaller-than-expected ETT size
-
Neuraxial anesthesia may be difficult due to spinal stenosis/deformity
-
Screen for pulmonary hypertension (from chronic apnea) - preoperative echocardiogram before major surgery
-
Miller's Anesthesia 10e, p. 9184
Comparison with Related FGFR3 Disorders
| Condition | FGFR3 Effect | Severity |
|---|---|---|
| Achondroplasia | Moderate gain-of-function (p.G380R) | Non-lethal; normal lifespan |
| Hypochondroplasia | Milder gain-of-function | Milder phenotype |
| Thanatophoric dysplasia | Stronger gain-of-function | Lethal at/near birth |
Recent Evidence Update
Three 2026 systematic reviews and meta-analyses on vosoritide confirm efficacy in increasing annualized height velocity in children with achondroplasia (PMIDs: 42026358, 41934413, 41424367). Long-term data on final adult height and safety beyond 2-3 years are still accumulating, so caution remains warranted.
Key pearl: Achondroplasia is compatible with a normal lifespan and normal intelligence. The most life-threatening complication in infancy is foramen magnum stenosis causing brainstem compression. The most disabling adult complication is lumbar spinal stenosis. Vosoritide is now an approved pharmacologic option to improve growth velocity in affected children.
Marfan syndrome
Finding Sources
Searching PubMed
Marfan syndrome FBN1 management guidelines
Reading File
Reading File
Reading File
Searching PubMed
Marfan syndrome aortic dissection treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive multi-source data. Here is the full synthesis:
Marfan Syndrome
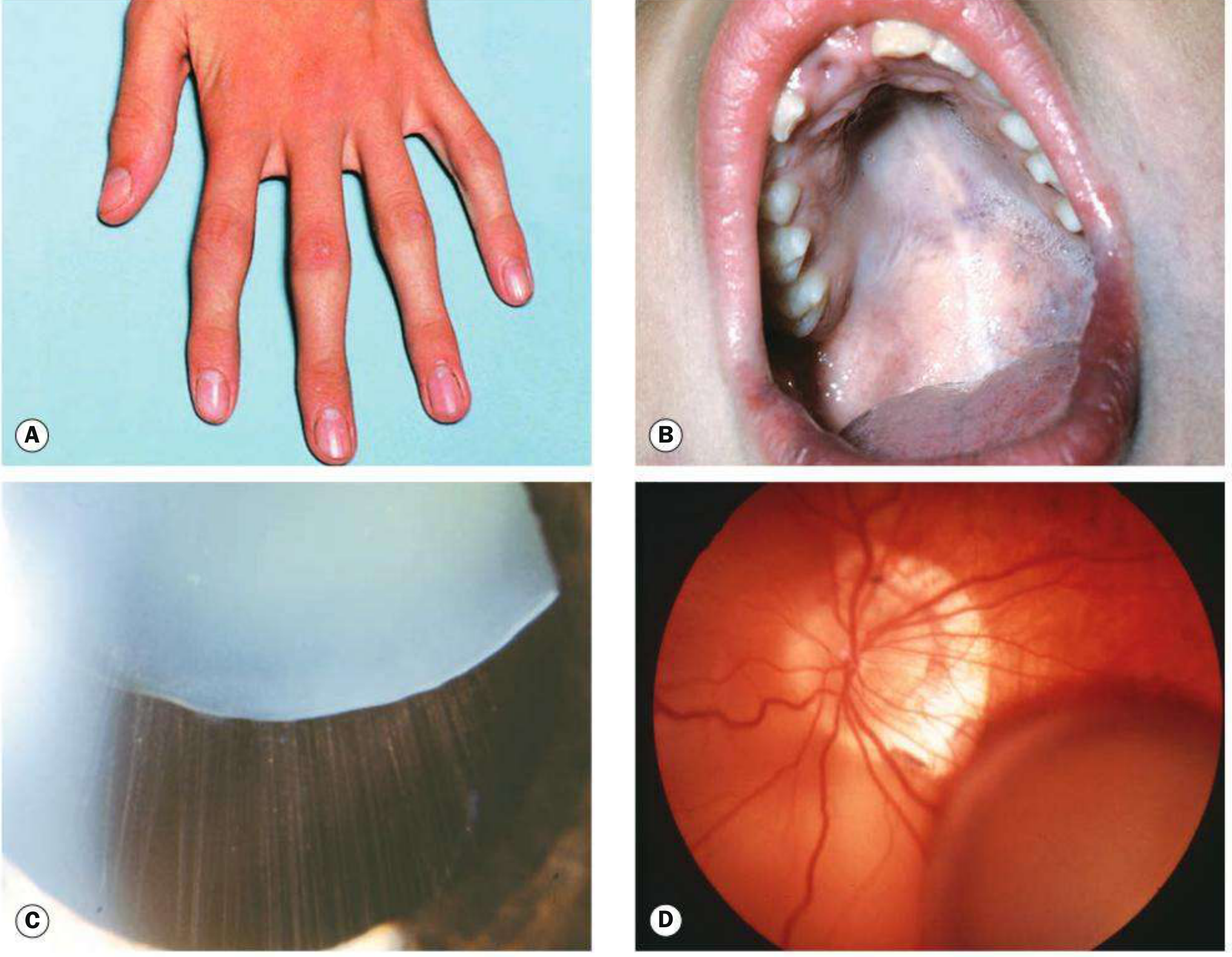
Overview
Marfan syndrome (MFS; OMIM #154700) is an autosomal dominant multisystem connective tissue disorder caused by mutations in the FBN1 gene. It affects primarily the cardiovascular, ocular, and musculoskeletal systems. The estimated prevalence is 1 in 3,000 to 5,000 individuals worldwide. Early mortality historically stemmed from aortic dissection; with modern surveillance and treatment, life expectancy now approximates that of the general population.
Genetics & Molecular Pathogenesis
| Feature | Detail |
|---|---|
| Gene | FBN1 (fibrillin-1), chromosome 15q21.1 |
| Gene size | 65 exons, 237 kb - one of the largest human genes |
| Mutation spectrum | ~2,000 variants reported; 66% single nucleotide variants, 20% small indels, 11% splice-site |
| Inheritance | Autosomal dominant, high penetrance |
| De novo rate | 15-30% (no family history) |
| Mechanism | Dominant negative - mutant fibrillin-1 incorporated into microfibrils disrupts normal microfibril assembly |
Fibrillin-1 and TGF-β
Fibrillin-1 is a large extracellular glycoprotein that forms 10-nm microfibrils. These microfibrils:
- Serve as scaffolds for elastin assembly in vascular walls, lung, and skin
- Provide tensile anchoring in non-elastic tissues (ciliary zonule of the eye, basement membranes)
- Sequester TGF-β, limiting its bioavailability
Loss of functional fibrillin-1 leads to excessive TGF-β signaling (via the SMAD2/3 pathway), which impairs vascular smooth muscle development and ECM integrity. This is the key mechanism for aortic complications - and the rationale for losartan (an AT1R blocker that inhibits TGF-β signaling) as therapy.
Notably, mutations in TGFBR2 (TGF-β type II receptor) cause the related Marfan syndrome type 2 (MFS2).
Genotype-phenotype correlation: Mutations in exons 24-32 ("neonatal region") cause early-onset, rapidly progressive, severe MFS with increased risk of ectopia lentis, aortic dilation, mitral anomalies, scoliosis, and reduced life expectancy.
- Robbins & Kumar Basic Pathology, p. 3356-3362
- Tietz Textbook of Laboratory Medicine, p. 2727-2728
- Firestein & Kelley's Rheumatology, p. 2342
Clinical Features
Cardiovascular (most life-threatening)
- Aortic root dilation at the sinus of Valsalva - present in ~50% of children, >80% of adults; progressive throughout life
- Aortic dissection - typically begins just above the coronary ostia and can extend the full length of the aorta
- Aortic regurgitation from dilation of the valve ring
- Mitral valve prolapse with regurgitation in 60-70% of patients; tricuspid valve involvement also occurs
- Proximal pulmonary artery dilation
- Cystic medionecrosis (fragmentation of elastic fibers in the tunica media) is the histologic finding
- Aortic rupture is the most common cause of death
Ocular
- Ectopia lentis (subluxation of the lens) in 50-80% of patients - typically bilateral and superotemporal (upward); appears by age 5; zonules usually intact so accommodation is preserved
- Myopia (>50%), the most common ocular feature
- Increased risk of retinal detachment, glaucoma, early cataracts
- Hypoplasia of the dilator pupillae (poor dilation with mydriatics)
Skeletal
- Tall stature with dolichostenomelia (disproportionately long limbs)
- Arachnodactyly (long fingers and toes)
- Arm span > height (ratio >1.05)
- Reduced upper body/lower extremity ratio
- High-arched ("gothic") palate with dental crowding
- Pectus excavatum or pectus carinatum (rib overgrowth)
- Scoliosis (~50% of patients); average onset age 10.5 years, rapid progression during adolescence
- Thoracic kyphosis, hindfoot deformity, pes planus
- Joint hypermobility (though elbows often have reduced extension)
- Reduced bone mineral density with increased fracture risk
- Protrusio acetabulae
Neurological
- Dural ectasia - enlargement of the caudal thecal sac occurring in up to 90% of patients (major diagnostic criterion); causes lumbosacral radiculopathy, low back pain, leg weakness/numbness, genital/rectal pain
- CSF leakage from ectatic dura → intracranial hypotension (positional headache)
- Sleep apnea (secondary to skeletal deformities)
- Stroke risk is mainly from cardiogenic emboli (prosthetic valves, AF)
Pulmonary
- Spontaneous pneumothorax (from apical blebs; score 2 in Ghent)
- Restrictive lung disease from scoliosis/kyphosis
Other
- Skin striae (without significant weight gain)
- Hernias (inguinal, umbilical)
Diagnosis - Revised Ghent Nosology (2010)
The diagnosis is based on a combination of family history, the two major cardinal features (aortic root aneurysm + ectopia lentis), systemic score, and FBN1 molecular testing.
Without family history - MFS is diagnosed if:
- Aortic root dilation (Z-score ≥ 2.0) + ectopia lentis, OR
- Aortic root dilation (Z-score ≥ 2.0) + pathogenic FBN1 variant, OR
- Aortic root dilation (Z-score ≥ 2.0) + systemic score ≥ 7, OR
- Ectopia lentis + pathogenic FBN1 variant known to cause aortic disease
With family history - MFS is diagnosed if:
- Ectopia lentis, OR
- Systemic score ≥ 7, OR
- Aortic root dilation (Z-score ≥ 2.0 in adults, ≥ 3.0 in children <20 years)
Systemic Score Table (Ghent)
| Feature | Score |
|---|---|
| Wrist AND thumb sign | 3 |
| Wrist OR thumb sign | 1 |
| Pectus carinatum | 2 |
| Pectus excavatum or chest asymmetry | 1 |
| Hindfoot deformity | 2 |
| Pes planus | 1 |
| Pneumothorax | 2 |
| Dural ectasia | 2 |
| Protrusio acetabulae | 2 |
| Reduced upper/lower segment ratio + increased arm span/height | 1 |
| Scoliosis or thoracolumbar kyphosis | 1 |
| Reduced elbow extension | 1 |
| 3 of 5 facial features (dolichocephaly, enophthalmos, down-slanting palpebral fissures, malar hypoplasia, retrognathia) | 1 |
| Skin striae | 1 |
| Myopia | 1 |
| Mitral valve prolapse | 1 |
| Threshold for Marfan diagnosis | ≥ 7 |
Clinical signs: Steinberg thumb sign (thumb extends beyond hypothenar border when enclosed in fist); Murdoch-Walker wrist sign (thumb overlaps 5th digit when encircling the opposite wrist).
- Firestein & Kelley's Rheumatology, p. 2343 (Table 107.3)
Management
Multidisciplinary Team
Cardiologist, ophthalmologist, orthopedic surgeon, geneticist, and (for reproductive-age women) obstetrician.
Cardiovascular Surveillance
- Annual transthoracic echocardiography to monitor aortic root diameter
- CT/MRI aortography for full aortic assessment
Pharmacotherapy
| Drug | Mechanism | Indication |
|---|---|---|
| Beta-blockers (atenolol, propranolol) | Reduce aortic wall stress by lowering heart rate and dP/dt | First-line; significantly slow aortic root dilation |
| Losartan (ARB) | Inhibits TGF-β signaling via AT1R blockade | Equal efficacy to atenolol in head-to-head RCT; used especially if beta-blockers not tolerated |
| Statins / tetracyclines | Inhibit matrix metalloproteinases (MMP-2, MMP-9) and ERK signaling | Adjunctive; reduce ECM degradation |
| Calcium channel blockers | - | AVOID - animal and clinical data show increased aneurysm expansion and rupture risk |
Surgical Indications (Aortic Root)
- Elective prophylactic aortic root replacement when aortic root reaches a critical diameter of 4.5-5.5 cm (elective repair has substantially better outcomes than emergency repair)
- Mitral valve replacement often performed concurrently
- Emergency surgery for dissection
Musculoskeletal
- Scoliosis: bracing first; surgical spinal fusion with rods if curvature >40-50 degrees
- Pectus deformities: surgery if interfering with pulmonary function
- Arthropathy from joint hypermobility: orthopedic intervention as needed
- Lens dislocation: surgical removal only if conventional vision correction is inadequate
Lifestyle
- Low-impact sports; avoid physical exhaustion, isometric exercise, and contact sports
- Avoid activities with risk for sudden blood pressure surges
- Restrict competitive athletics
Special Populations
- Pregnancy: increased risk of aortic dissection, especially with pre-existing aortic dilation; close monitoring and multidisciplinary management required
- Pediatric patients: may carry "potential MFS" diagnosis requiring periodic re-evaluation as features emerge with age
Differential Diagnosis
| Condition | Key Distinguishing Features |
|---|---|
| Homocystinuria (CBS deficiency) | AR inheritance; lens dislocation is inferior or lateral (vs. superior in MFS); intellectual disability; venous/arterial thrombosis; no progressive aortic root dilation; elevated homocysteine |
| Loeys-Dietz syndrome | TGFBR1/2, SMAD3, TGFB2 mutations; no ectopia lentis; may have bifid uvula, craniosynostosis, hypertelorism; aggressive aortic disease at smaller diameters |
| MASS syndrome | MFS features (Mitral, Aorta, Skeletal, Skin) but no ectopia lentis and no significant aortic dilation |
| Weill-Marchesani syndrome | Conceptually the "converse" of MFS - short stature, brachydactyly, microspherophakia; AD form also involves FBN1 |
| Ehlers-Danlos syndrome | Joint hypermobility predominates; skin hyperextensibility; different molecular basis |
Prognosis
- Untreated life expectancy historically ~32 years (death from aortic catastrophe)
- With beta-blockade and regular surveillance: life expectancy approaches that of the general population
- Elective aortic surgery has dramatically better outcomes than emergency repair
- Wide phenotypic variability - some patients mildly affected, others severely affected even with the same mutation
Key pearls:
- The two cardinal features driving diagnosis are aortic root aneurysm and ectopia lentis (bilateral, superotemporal)
- Pathogenesis is via excess TGF-β signaling, not just structural connective tissue weakness - which explains why ARBs (losartan) are therapeutic
- Calcium channel blockers are contraindicated in MFS
- Dural ectasia (90%) is a highly sensitive but underrecognized feature
- In homocystinuria (the key mimic), lens displacement is inferior/lateral, not superior - and there is no aortic root dilation
- Robbins & Kumar Basic Pathology, p. 3356-3371
- Tietz Textbook of Laboratory Medicine, p. 2727-2730
- Firestein & Kelley's Rheumatology 2-Volume Set, p. 2342-2344
- Bradley and Daroff's Neurology, p. 2757-2769
- Kanski's Clinical Ophthalmology 10th Ed, p. 355-356
Fetal alcohol syndrome
Finding Sources
Searching PubMed
fetal alcohol syndrome prenatal alcohol exposure
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Excellent - I now have rich, comprehensive data from multiple authoritative sources. Here is the full synthesis:
Fetal Alcohol Syndrome (FAS) and Fetal Alcohol Spectrum Disorders (FASD)
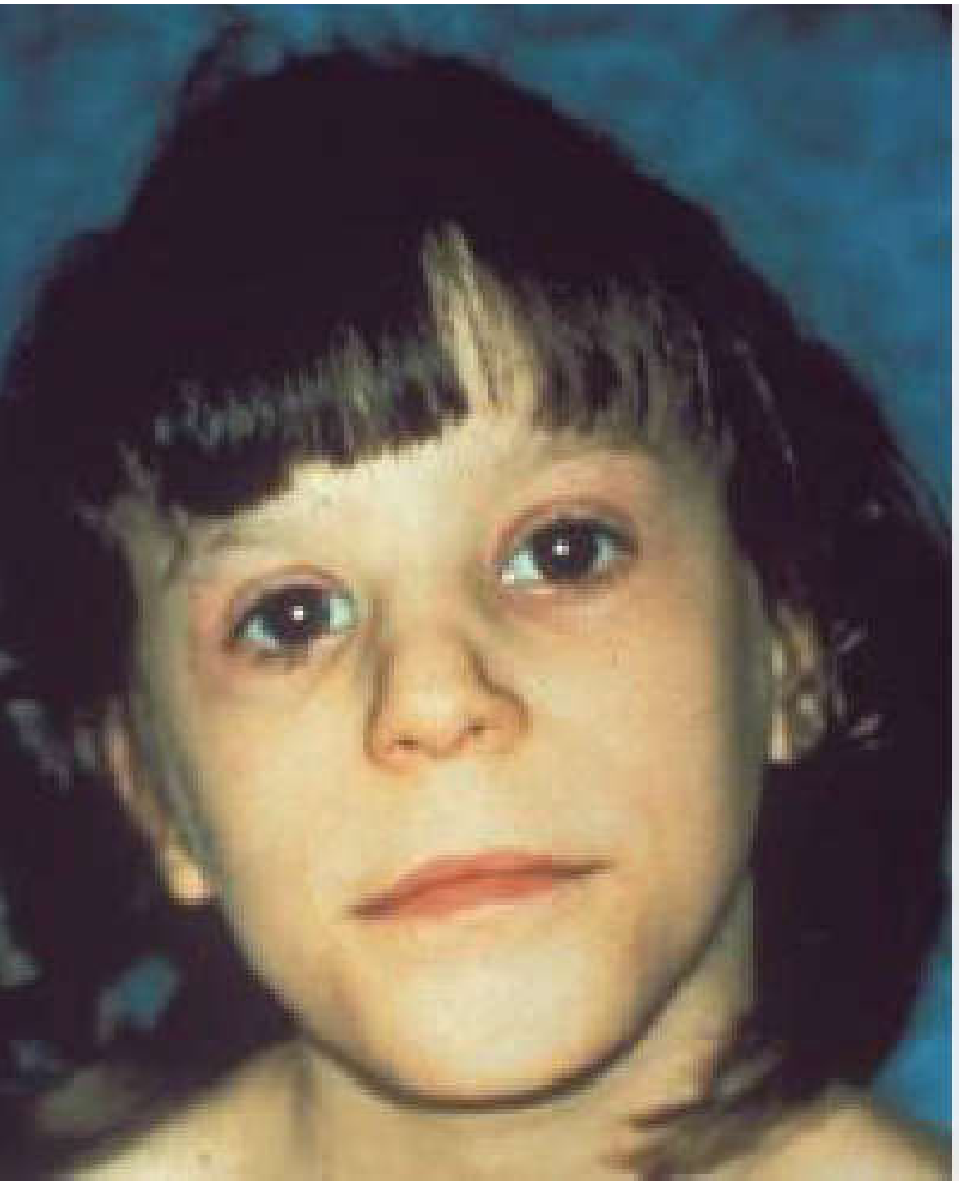
Overview & Terminology
Fetal alcohol syndrome (FAS) is the severe end of a spectrum of conditions caused by prenatal alcohol exposure. The umbrella term fetal alcohol spectrum disorder (FASD) encompasses all alcohol-related birth defects and neurodevelopmental abnormalities. FASD is considered the most common nongenetic, entirely preventable cause of intellectual disability.
-
Incidence of FAS: 2-9 per 1,000 births
-
Incidence of FASD (all spectrum): 24-48 per 1,000 births; active surveillance in US first-graders found rates as high as 31-98.5 per 1,000
-
The CDC estimates up to 1 in 20 US school-age children may have FASDs
-
Langman's Medical Embryology, p. 155
-
Creasy & Resnik's Maternal-Fetal Medicine, p. 1555-1557
Pathophysiology
Alcohol as a Teratogen
Ethanol freely and rapidly crosses the placenta, reaching fetal concentrations equal to maternal blood levels. The fetal liver has little or no alcohol dehydrogenase (ADH) activity, so the fetus depends on maternal and placental enzymes for alcohol elimination - resulting in prolonged fetal exposure.
Key Mechanisms of Injury
| Mechanism | Detail |
|---|---|
| Apoptotic neurodegeneration | Alcohol triggers programmed death of neurons during synaptogenesis (begins ~6th month of gestation, extends through early postnatal life); the most vulnerable period for CNS injury |
| Aberrant neuronal/glial migration | Disrupted migration during brain development |
| Sonic Hedgehog (SHH) suppression | Alcohol downregulates SHH signaling by interfering with SHH-cholesterol binding, causing abnormal brain development and death of neural crest cells responsible for craniofacial structures |
| Inhibition of neurite outgrowth | Demonstrated in tissue culture systems |
| Acetaldehyde toxicity | The primary ethanol metabolite can independently damage developing tissues |
The effects are not simply due to malnutrition - infants born to malnourished but sober mothers (e.g., during World War II famine) were small and premature but lacked the FAS malformation pattern.
- Katzung's Basic and Clinical Pharmacology 16th Ed, p. 628
- Adams and Victor's Principles of Neurology 12th Ed, p. 1200
- Langman's Medical Embryology, p. 155-156
Clinical Features
The Three Diagnostic Domains of FAS
1. Facial Dysmorphology (3 Key Features)
- Short palpebral fissures (shortened eye opening)
- Smooth philtrum (absent or indistinct groove between nose and upper lip)
- Thin vermilion border (thin upper lip)
Additional facial features: maxillary hypoplasia, micrognathia, depressed nasal bridge, short nose, flat midface, epicanthal folds. The ear helix may show a "railroad track" configuration. These facial stigmata become less distinctive after puberty.
2. Growth Deficiency
- Intrauterine growth restriction (IUGR)
- Weight and length <10th percentile
- Microcephaly (head circumference <10th percentile) - persistent throughout childhood
- Slow head circumference growth is a consistent finding throughout infancy and childhood
3. Central Nervous System Abnormalities
Structural: Reduced overall brain size on fMRI; specific size reductions in the basal ganglia and cerebellum; impaired or absent corpus callosum (agenesis/hypoplasia); microcephaly; rarely holoprosencephaly
Neurologic: Hypotonia, poor coordination, tremulousness in neonates (mimicking alcohol withdrawal but persisting), seizures
Functional/Neurocognitive:
- Average IQ ~72 in full FAS; ~80 in heavy prenatal exposure without FAS
- The majority do not have IQ <70, so many do not meet formal intellectual disability criteria
- Deficits in verbal learning/memory (predominantly encoding), executive function (planning, working memory, response inhibition, set-shifting, concept formation), visual-spatial reasoning, attention, and academic achievement (reading, spelling, mathematics)
Behavioral & Psychiatric Features
- ADHD - most common psychiatric comorbidity; prevalence 44-96% in FASD
- Impulsivity, rule-breaking, anxiety, poor social interactions
- Oppositional defiant disorder (ODD) and conduct disorder
- Deficits in socialization and communication that worsen with age (unlike idiopathic ADHD)
- In adolescence and adulthood: legal problems, substance abuse disorders, psychiatric disorders, elevated suicide risk
Other Features
-
Minor cardiac anomalies (cardiac septal defects - often self-closing)
-
Joint anomalies, flexion deformities of fingers, limited range of joint motion
-
Longitudinally oriented palmar creases ("hockey-stick" crease)
-
Cleft lip and palate (more frequent than in general population)
-
Anomalous external genitalia
-
Poor sucking and sleeping in neonates; irritability, restlessness, hyperactivity
-
Adams and Victor's Principles of Neurology, p. 1199-1200
-
Goldman-Cecil Medicine, p. 249-251
-
Kaplan & Sadock's Comprehensive Textbook of Psychiatry, p. 2041-2044
FASD Spectrum - Diagnostic Framework (Hoyme et al. 2016)
| Diagnosis | Confirmed Prenatal Alcohol Exposure | Facial Anomalies | Growth Deficiency | CNS Abnormalities | Neurobehavioral Impairment |
|---|---|---|---|---|---|
| FAS | Not required | Required | Required | Required | Required |
| Partial FAS (with documented exposure) | Required | Required | Not required | Not required | Required |
| Partial FAS (without documented exposure) | Not required | Required | ≥1 required | - | Required |
| ARND (Alcohol-Related Neurodevelopmental Disorder) | Required | Not required | Not required | Not required | Required |
| ND-PAE (DSM-5 appendix) | Required | Not required | Not required | Not required | Required |
Key point: Full FAS diagnosis does not require confirmed maternal alcohol exposure history - the facial + growth + CNS combination is specific enough to diagnose without it.
ARND captures the large population affected neurodevelopmentally but lacking physical dysmorphism - these children are often missed.
- Kaplan & Sadock's, Table 2.13-2, p. 2041
ND-PAE (DSM-5 Conditions for Further Study)
Requires more than minimal prenatal alcohol exposure PLUS all three:
- Neurocognitive impairment (≥1 of: intellectual, executive function, learning, memory, visual-spatial)
- Self-regulation impairment (≥1 of: mood/behavioral regulation, attention, impulse control)
- Adaptive functioning impairment (≥2 of: communication, social interaction, daily living skills, motor skills)
Timing of Exposure
| Trimester | Predominant Effect |
|---|---|
| First | Facial dysmorphia, major structural brain malformations (organogenesis) |
| Second | Growth restriction (birth length) |
| Third | Birth weight reduction; greatest vulnerability window for apoptotic neurodegeneration (synaptogenesis) |
Binge drinking (>5 drinks per sitting) at any critical developmental stage increases risk of birth defects including orofacial clefts. No safe threshold of alcohol consumption in pregnancy has been established. Even mild-to-moderate intake may cause harm. Risk is not solely determined by quantity or timing - twin studies show identical prenatal exposure can produce markedly different FASD outcomes, suggesting genetic modifier effects.
Comparison: FASD vs. Idiopathic ADHD
Children with FASD are often misdiagnosed as having ADHD alone. Key distinguishing points:
| Feature | FASD | Idiopathic ADHD |
|---|---|---|
| IQ | Lower overall (~72-80) | Typically in normal range |
| Attention deficit | Impaired shifting + encoding | Primarily focusing/sustaining |
| Socialization | Arrested development, worsens with age | Does not worsen with age |
| Verbal encoding | More impaired | Less impaired |
| Adaptive behavior | Broader impairment across all domains | Less impaired |
| Overall severity | Greater cognitive-behavioral impairment | Less impaired |
Prevention
- Complete alcohol abstinence throughout pregnancy is the only safe approach
- No safe level of alcohol, no safe type of drink, and no safe time during pregnancy has been identified
- Breastfeeding: alcohol concentrations in breast milk mirror maternal blood; AAP advises maximum one drink, then waiting ≥2 hours before nursing; alcohol during lactation is associated with dose-dependent reductions in abstract reasoning at 6-7 years
Management
FASD has no cure. Management is supportive and multidisciplinary:
- Early intervention is associated with improved long-term outcomes; early diagnosis is therefore important
- Neurodevelopmental support: special education, speech/language therapy, occupational therapy
- Behavioral interventions targeting executive function, social skills, and adaptive behavior
- Pharmacotherapy: ADHD medications may reduce attentional symptoms; however, children with FASD and comorbid ADHD have greater impairment than ADHD alone and may respond differently; no FASD-specific pharmacotherapy exists
- Psychiatric management of comorbid ODD, conduct disorder, anxiety, depression
- Family support and psychoeducation
- In 2025, the first evidence-based guideline for interventions in FASD was published (Strieker et al., Neuropediatrics 2025 - PMID 40015329)
Recent Evidence Update (2024-2025)
- A 2024 systematic review and meta-analysis (Akison et al., BMC Med - PMID 39407296) confirmed dose-dependent associations between prenatal alcohol exposure and reduced physical size, dysmorphology, and neurodevelopmental outcomes
- A 2024 meta-analysis (Clark et al., J Affect Disord - PMID 38718944) documented high prevalence of co-occurring psychiatric diagnoses in alcohol-exposed individuals
- A 2024 systematic review (Leung et al., Alcohol - PMID 38097146) highlighted the cerebellum as a particularly sensitive structure in FASD - consistent with clinical findings of motor incoordination
- The first evidence-based intervention guideline was published in 2025 (PMID 40015329)
Key pearls:
- The three cardinal facial features are short palpebral fissures + smooth philtrum + thin upper lip - and two of three are sufficient for the facial criterion
- Full FAS does not require confirmed alcohol exposure history - the clinical phenotype alone is diagnostic
- The corpus callosum and cerebellum are the brain structures most consistently affected
- FASD children perform worse than idiopathic ADHD on cognitive measures - this distinction matters for management
- There is no safe level of alcohol in pregnancy - total abstinence is the only recommendation
- Adams and Victor's Principles of Neurology 12th Ed, p. 1199-1200
- Katzung's Basic and Clinical Pharmacology 16th Ed, p. 628
- Kaplan & Sadock's Comprehensive Textbook of Psychiatry, p. 2040-2045
- Langman's Medical Embryology, p. 155-156
- Creasy & Resnik's Maternal-Fetal Medicine, p. 1555-1561
Fetal alcohol syndrome
It looks like you just asked about fetal alcohol syndrome - I covered it in detail in my previous response! Here's a quick summary of the key points:
FAS / FASD - Summary
Definition: FAS is the severe end of the fetal alcohol spectrum disorder (FASD) umbrella - the leading preventable, nongenetic cause of intellectual disability.
Three diagnostic domains of FAS:
- Facial dysmorphology - short palpebral fissures, smooth philtrum, thin upper lip
- Growth deficiency - IUGR, weight/length/head circumference <10th percentile
- CNS abnormalities - microcephaly, corpus callosum agenesis, basal ganglia/cerebellar reduction, neurocognitive deficits
Mechanism: Alcohol crosses the placenta freely; fetal liver lacks ADH; teratogenicity via apoptotic neurodegeneration (especially during synaptogenesis), aberrant neuronal migration, and SHH pathway suppression.
No safe level of alcohol in pregnancy has been established - complete abstinence is recommended.
Management is supportive: early intervention, special education, behavioral therapies, ADHD pharmacotherapy for attentional symptoms.
If you were looking for something more specific - such as a particular aspect of FAS (e.g., neuroimaging findings, differential diagnosis, management details, or the FASD spectrum criteria table) - just let me know and I'll focus on that.
This is a shared conversation. Sign in to Orris to start your own chat.